4. Materials and Methods
General Methods. HPLC grade solvents and reagents were obtained from commercial suppliers and were used without further purification. Chrysin (
1), compound
2, and 5,7-dihydroxychromen-4-one (
13) were synthesized according to the methodologies previously described by us.
11,12 LCMS experiments were performed in a column XBridge C18 3.5u 2.1 × 50 mm at 1.2 mL/min and 50 °C; 10 mM ammonium bicarbonate pH 9/ACN, gradient 10 > 95% ACN in 1.5 min + 0.5 min hold. Reactions affording compounds
37 and
17 were followed by TLC, carried out on aluminum sheets (20 × 20 cm) coated with silica gel 60 F-254, 0.2 mm thick (Merck, Darmstadt, Germany) with detection by charring with 10% H
2SO
4 in ethanol. Flash column chromatography was performed using CombiFlash
® Rf200 (Teledyne Isco, Lincoln, CA, USA). Preparative HPLC was performed in a Gilson apparatus using either Phenomenex Gemini NX, C18, 5 μm 30 × 100 mm or Phenomenex Gemini NX, C18, 10 μm 50 × 150 mm columns. NMR spectra for compound characterization were recorded on a Bruker AV III HD Nanobay spectrometer running at 400.13 MHz equipped with a room temperature 5 mm BBO Smartprobe. Chemical shifts are expressed in δ (ppm) and the proton coupling constants
J in Hertz (Hz). NMR data were assigned using appropriate COSY, DEPT, HMQC, and HMBC spectra (representative examples are provided in the
Supporting information appendix). Optical rotations were measured with a Perkin–Elmer 343 polarimeter. Melting points were measured using a Stuart SMP30 melting point apparatus. High-resolution mass spectra of final compounds were acquired on a Bruker Daltonics HR QqTOF Impact II mass spectrometer (Billerica, MA, USA). The nebulizer gas (N
2) pressure was set to 1.4 bar, and the drying gas (N
2) flow rate was set to 4.0 L/minute at a temperature of 200 °C. The capillary voltage was set to 4500 V and the charging voltage was set to 2000 V. Tested compounds have ≥ 95% purity as determined by LCMS. Synthesis of intermediate compounds
23–
31 and
35–
42 is reported in
Supplementary Materials, together with their LCMS data, physical and NMR data.
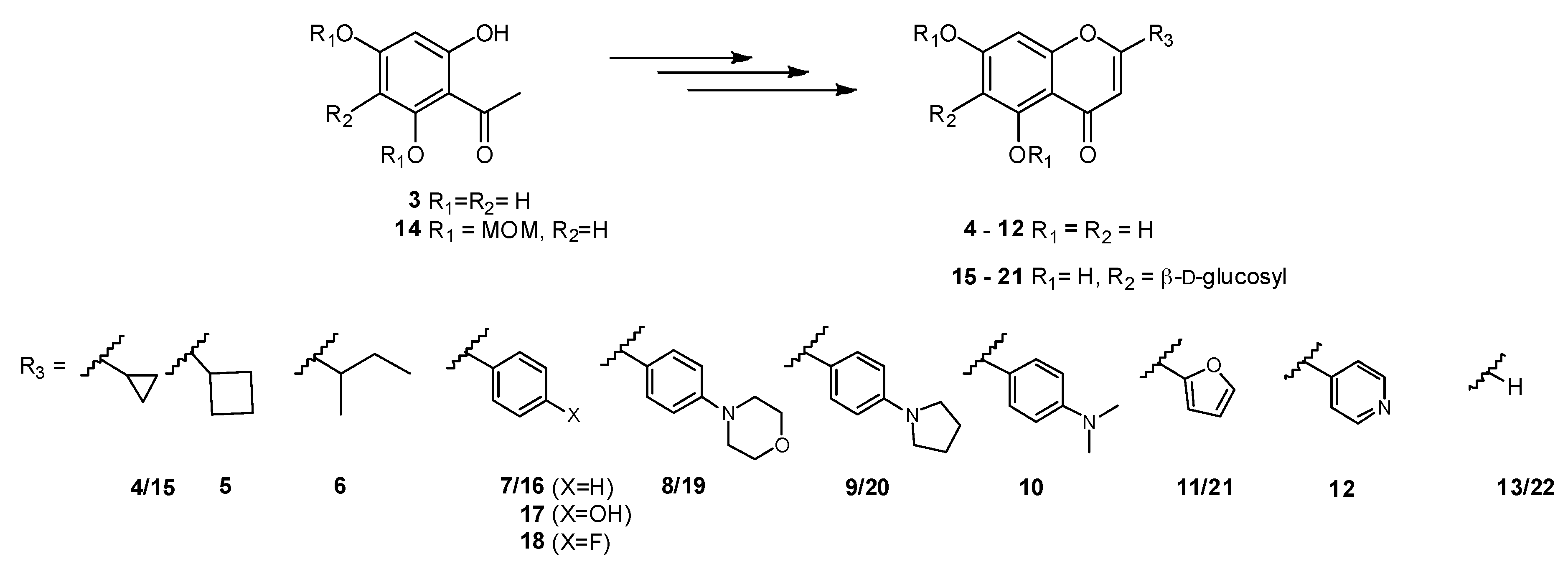
General procedure for the synthesis of non-glycosylated chalcones/flavones. Each compound 23–30 was dissolved in dry pyridine (0.248 mmol in 7.33 mL). Then, catalytic amounts of I2 (0.087 mmol, 0.35 eq.) were added and the mixture was stirred under reflux for 24 h–72 h. All reactions were followed by LCMS. Once the starting material was fully consumed, the mixture was allowed to reach room temperature and the pyridine was co-evaporated with toluene under reduced pressure. The residue was resuspended in dichloromethane, washed first with a saturated solution of sodium thiosulfate, and then with brine. The flavone was extracted with dichloromethane (3 × 30 mL), dried over MgSO4, and the solution filtered and concentrated under vacuum. The residue was then resuspended in ethanol (15 mL) and p-TsOH (12% in AcOH, 0.1 mL) was added. The reaction was stirred under reflux for 2–24 h. After having reached completion by LCMS, the solvent was evaporated under vacuum and the residue purified using the most adequate purification method(s) to afford compounds 4–12.
2-Cyclopropyl-5,7-dihydroxy-4H-chromen-4-one (4). Purified by preparative HPLC. Reaction yield over two steps: 53%; LCMS: RT = 0.52 min, m/z = 219.0 [M + H]+ (high pH method); white solid; m.p. = 201.2–202.9 °C. 1H NMR (MeOD) δ (ppm) 6.26 (d, 1H, Jmeta = 2.1 Hz, H-8), 6.17 (d, 1H, Jmeta = 2.1 Hz, H-6), 6.10 (s, 1H, H-3), 2.02–1.96 (m, 1H, H-1′), 1.18–1.09 (m, 4H, H-2′, H-3′). 13C NMR [MeOD] δ (ppm) 181.5 (C-4), 173.5 (C-2), 166.0 (C-7), 163.4 (C-5), 159.6 (C-8a), 106.5 (C-3), 100.2 (C-6), 95.0 (C-8), 15.4 (C-1′), 9.5 (C-2′, C-3′). HRMS-ESI (m/z): [M + H]+ calcd for C12H11O4 219.0652, found 219.0642.
2-Cyclobutyl-5,7-dihydroxy-4H-chromen-4-one (5). Purified by preparative HPLC. Reaction yield over two steps: 85%; LCMS: RT = 0.72 min, m/z = 233.0 [M + H]+ (high pH method); white solid; m.p. = 200.3–201.5 °C. 1H NMR (MeOD) δ (ppm) 6.34 (d, 1H, Jmeta = 2.1 Hz, H-8), 6.18 (d, 1H, Jmeta = 2.1 Hz, H-6), 6.03 (s, 1H, H-3), 3.53 (td, 1H, J = 8.0 Hz, H-1′), 2.37–2.31 (m, 4H, H-2′, H-4′), 2.17–2.06 (m, 1H, H-3′a), 1.99–1.91 (m, 1H, H-3′). 13C NMR [(CD3)OD] δ (ppm) 183.4 (C-4), 174.1 (C-2), 166.0 (C-7), 163.3 (C-5), 159.9 (C-8a), 106.5 (C-3), 100.0 (C-6), 94.9 (C-8), 39.5 (C-1′), 27.5 (C-2′, C-4′), 19.0 (C-3′). HRMS-ESI (m/z): [M + H]+ calcd for C13H13NO4 233.0808, found 233.0804.
5,7-Dihydroxy-2-(1-methylpropyl)-4H-chromen-4-one (6). Purified by preparative HPLC. Reaction yield over three steps: 67%; LCMS: RT = 1.10 min, m/z = 235.0 [M + H]+ (low pH method); brown solid; m.p. = 192.5–193.8 °C. 1H NMR (MeOD) δ (ppm) 6.33 (d, 1H, Jmeta = 2.2 Hz, H-8), 6.20 (d, 1H, Jmeta = 2.2 Hz, H-6), 6.05 (s, 1H, H-3), 2.65 (sextet, 1H, J1′-1″~1′-2′ = 7.4 Hz, H-1′), 1.80–1.59 (m, 2H, H-2′), 1.30 (d, 3H, J1″-1′ = 7.4 Hz, H-1″), 0.95 (t, 3H, J3′-2′ = 7.4 Hz, H-3′). 13C NMR [MeOD] δ (ppm) 184.1 (C-4), 175.8 (C-2), 166.0 (C-7), 163.3 (C-5), 159.9 (C-8a), 107.6 (C-3), 105.4 (C-4a), 100.0 (C-6), 94.8 (C-8), 41.7 (C-1′), 28.6 (C-2′), 18.2 (C-1″), 11.9 (C-3′). HRMS-ESI (m/z): [M + H]+ calcd for C13H15O4 235.0965, found 235.0962.
4′-Fluoro-5,7-dihydroxyflavone (7). Purified by preparative HPLC. Reaction yield over two steps: 38%; LCMS: RT = 1.10 min, m/z = 273.0 [M + H]+ (low pH method); white solid; m.p. = 264.8–265.5 °C. 1H NMR [MeOD] δ (ppm) 8.05 (dd, 2H, Jortho = 8.9 Hz, J2′-F=6′-F = 5.2 Hz, H-2′, H-6′), 7.31 (t, 2H, Jortho~J3′-F=5′-F = 8.7 Hz, H-3′, H-5′), 6.73 (s, 1H, H-3), 6.50 (d, 1H, Jmeta = 2.2 Hz, 1H, H-8), 6.24 (d, 1H, Jmeta = 2.2 Hz, H-6). 13C NMR [MeOD] δ (ppm) 183.8 (C-4), 166.3 (C-7), 164.7 (C-5), 163.3 (d, JC-F = 259.3 Hz, C-4′), 159.5 (C-8a), 130.1 (d, JC-F = 9.0 Hz, C-2′, C-6′), 127.6 (C-1′), 117.3 (d, JC-F = 22.5 Hz, C-3′, C-5′), 106.0 (C-3, C-4a), 100.3 (C-6), 95.2 (C-8). HRMS-ESI (m/z): [M + H]+ calcd for C15H10FO4 273.0558, found 273.0554.
5,7-Dihydroxy-4′-(morpholin-4-yl)flavone (8). Purified by preparative HPLC. Reaction yield over two steps: 68%; LCMS: RT = 1.07 min, m/z = 340.0 [M + H]+ (low pH method); orange solid; m.p. = 234.0–235.6 °C. 1H NMR (MeOD) δ (ppm) 7.87 (d, 2H, Jortho = 9.0 Hz, H-2′ and H-6′), 7.06 (d, 2H, H-3′ and H-5′), 6.58 (s, 1H, H-3), 6.45 (d, 1H, Jmeta = 2.2 Hz, H-8), 6.21 (d, 1H, Jmeta = 2.1 Hz, H-6), 3.86–3.83 (m, 4H, NCH2CH2O), 3.34 (NCH2CH2O, overlapped with metanol-d4 peak). 13C NMR (MeOD) δ (ppm) 183.8 (C-4), 165.9 (C-2), 163.7 (C-7 and C-5), 159.4.0 (C-8a), 155.3 (C-4′), 128.9 (C-2′, C-6′), 121.8 (C-1′), 115.4 (C-3′, C-5′), 105.3 (C-4a), 103.3 (C-3), 100.1 (C-6), 95.0 (C-8), 67.7 (NCH2CH2O), 48.1 (NCH2CH2O). HRMS-ESI (m/z): [M + H]+ calcd for C19H18NO5 340.1179, found 340.1175.
5,7-Dihydroxy-4′-(pyrrolidin-1-yl)flavone (9). Purified by preparative HPLC. Reaction yield over two steps: 95%; LCMS: RT = 1.08 min, m/z = 324.0 [M + H]+ (high pH method); orange solid; m.p. = 282.4–283.6 °C. 1H NMR [(CD3)2CO] δ (ppm) 7.89 (d, 2H, Jortho = 9.0 Hz, H-2′ and H-6′), 6.70 (s, 1H, H-3), 6.65 (d, 2H, H-3′ and H-5′), 6.46 (d, 1H, Jmeta = 2.2 Hz, H-8), 6.16 (d, 1H, Jmeta = 2.2 Hz, H-6), 3.35–3.33 (m, NCH2CH2), 2.00–1.97 (m, 4H, NCH2CH2). 13C NMR [(CD3)2CO] δ (ppm) 181.5 (C-4), 164.6 (C-2), 163.9 (C-7), 161.5 (C-5), 157.2 (C-8a), 150.1 (C-4′), 128.0 (C-2′, C-6′), 116.0 (C-1′), 111.7 (C-3′, C-5′), 103.6 (C-4a), 100.9 (C-3), 98.7 (C-6), 93.9 (C-8), 47.4 (NCH2CH2), 25.0 (NCH2CH2). HRMS-ESI (m/z): [M + H]+ calcd for C19H18NO4 324.1230, found 324.1225.
4′-Dimethylamino-5,7-dihydroxyflavone (10). Purified by preparative HPLC. Reaction yield over two steps: 54%; LCMS: RT = 1.12 min, m/z = 298.0 [M + H]+ (low pH method); orange solid; m.p. = 291.3–292.7 °C. 1H NMR [(CD3)2SO] δ (ppm) 13.11 (s, 1H, OH-5), 7.88 (d, 2H, Jortho = 9.0 Hz, H-2′ and H-6′), 6.80 (d, 2H, H-3′ and H-5′), 6.71 (s, 1H, H-3), 6.46 (d, 1H, Jmeta = 2.1 Hz, H-8), 6.17 (d, 1H, Jmeta = 2.1 Hz, H-6), 3.03 [s, 6H, N(CH3)2]. 13C NMR [(CD3)2SO] δ (ppm) 181.5 (C-4), 164.3 (C-2), 163.8 (C-7), 161.4 (C-5), 157.2 (C-8a), 152.6 (C-4′), 127.8 (C-2′, C-6′), 116.5 (C-1′), 111.6 (C-3′, C-5′), 103.6 (C-4a), 101.2 (C-3), 98.6 (C-6), 93.8 (C-8), 39.6 [N(CH3)2]. HRMS-ESI (m/z): [M + H]+ calcd for C17H16NO4 298.1074, found 298.101.
2-(Furan-2-yl)-5,7-dihydroxy-4H-chromen-4-one (11). Purified by preparative HPLC. Reaction yield over two steps: 59%; LCMS: RT. = 0.99 min, m/z = 298.0 [M + H]+ (low pH method); white solid; m.p. = 239.3–240.8 °C. 1H NMR [(CD3)2CO] δ (ppm) 12.87 (s, 1H, OH-5), 7.92 (d, 1H, J5′-4′ = 1.6 Hz, H-5′), 7.34 (d, 1H, J4′-3′ = 3.5 Hz, H-3′), 6.77 (dd, 1H, J5′-4′ = 3.5 Hz, J3′-4′ = 1.8 Hz, H-4′), 6.49–6.48 (m, 2H, H-3, H-8), 6.27 (d, 1H, Jmeta = 1.8 Hz, H-6). 13C NMR [(CD3)2CO] δ (ppm) 182.7 (C-4), 164.9 (C-2), 156.3 (C-7)*, 163.1 (C-5), 158.0 (C-8a)*, 147.8 (C-5′), 146.9 (C-2′), 114.8 (C-3′), 113.9 (C-4′), 104.0 (C-4a), 103.9 (C-3), 100.1 (C-6), 95.0 (C-8). HRMS-ESI (m/z): [M + H]+ calcd for C13H19O5 245.0444, found 245.0438. *Permutable signals.
5,7-Dihydroxy-2-(pyridin-4-yl)-4H-chromen-4-one (12). Purified by preparative HPLC. Reaction yield over three steps: 87%; LCMS: RT = 0.43 min, m/z = 256.0 [M + H]+ (low pH method);: orange oil. 1H NMR (MeOD) δ (ppm) 8.79 (d, 2H, H-3′, H-5′), 8.37 (d, 2H, Jortho = 6.6 Hz, H-2′, H-6′), 6.74 (s, 1H, H-3), 6.30 (d, 1H, Jmeta = 1.8 Hz, H-8), 6.10 (d, 1H, Jmeta = 1.8 Hz, H-6). 13C NMR (MeOD) δ (ppm) 180.7 (C-4), 171.1 (C-2), 169.8 (C-7), 161.0 (C-5), 155.6 (C-8a), 151.6 (C-1′), 143.3 (C-3′, C-5′), 128.1 (C-2′, C-6′), 103.4 (C-3), 103.2 (C-4a), 99.6 (C-6), 92.7 (C-8). HRMS-ESI (m/z): [M + H]+ calcd for C14H10NO4 256.0604, found 256.0600.
General procedure for the synthesis of C-glucosylflavones. Each C-glucosylchalcone 35–41 was dissolved in dry pyridine (0.172 mmol in 5.11 mL). Then, catalytic amounts of I2 (0.060 mmol, 0.35 eq.) were added and the mixture was stirred under reflux for 48–72 h. All reactions were followed by LCMS. Once the starting material was fully consumed, the mixture was allowed to reach room temperature and the pyridine was co-evaporated with toluene under reduced pressure. The residue was resuspended in dichloromethane, washed first with a saturated solution of sodium thiosulfate, and then with brine. The flavone was extracted with dichloromethane (3 × 30 mL), dried over MgSO4, and the solution filtered and concentrated under vacuum. The residue was then resuspended in extra dry dichloromethane (7.10 mL) and stirred at −78 °C under N2 saturated atmosphere. A 1 M solution of BBr3 in dichloromethane (1.72 mL, 1.72 mmol, 10 eq.) was added in a dropwise manner over 5 min, and the reaction stirred for 2–4 h. After having reached completion by LCMS, the reaction was quenched with a 1:1 mixture of dichloromethane/methanol (ca. 15 mL) and the reaction was stirred for approximately 20 min at room temperature. The solvent was evaporated under vacuum and the residue purified using the most adequate purification method(s) to afford compounds 15–21.
2-Cyclopropyl-8-(β-d-glucopyranosyl)-5,7-dihydroxy-4-chromen-4-one (15). Purified by preparative HPLC. Reaction yield over two steps: 37%; LCMS: RT = 0.51 min, m/z = 381.0 [M + H]+ (low pH method); yellowish oil. 1H NMR (MeOD) δ (ppm) 6.23 (s, 1H, H-6), 6.20 (s, 1H, H-3), 4.86 (H-1″, overlapped with the methanol-d6 water peak), 3.92–3.87 (m, 2H, H-2″, H-6″a), 3.75–3.64 (m, 1H, H-6″b), 3.47–3.42 (m, 3H, H-3″, H-4″, H-5″), 2.06–2.00 (m, 1H, H-1′), 1.46–1.43 (m, 1H, H-2′a)*, 1.28–1.22 (m, 1H, H-3′a)*, 1.18–1.09 (m, 2H, H-2′b, H-3′b). 13C NMR (MeOD) δ (ppm) 183.6 (C-4), 173.3 (C-2), 164.4 (C-7), 162.7 (C-5), 160.4 (C-8a), 106.8 (C-8), 105.5 (C-3), 104.9 (C-4a), 99.3 (C-6), 82.8 (C-5″), 80.1 (C-3″), 74.9 (C-1″), 73.3 (C-2″), 72.6 (C-4″), 63.4 (C-6″), 15.7 (C-1′), 10.0, 9.6 (C-2′, C-3′). *Permutable signals. HRMS-ESI (m/z): [M + H]+ calcd for C18H21O9 381.1180, found 381.1176.
8-(β-d-Glucopyranosyl)-5,7-dihydroxyflavone (16). Purified by preparative HPLC, followed by Isolute SCX-2 column chromatography (Biotage). Reaction yield over two steps: 88%; LCMS: RT = 0.49 min, m/z = 414.80 [M − H]− (high pH method); yellow solid; m.p. = 188.1–189.2 °C. 1H NMR (MeOD) δ (ppm) 8.13, 8.03 (d, 2H, Jortho = 7.1 Hz, H-2′ and H-6′)*, 7.58–7.54 (m, 3H, H-3′, H-4′ and H-5′), 6.75 (s, 1H, H-3), 6.30 (s, 1H, H-6), 5.00 (d, 1H, J1″-2″ = 9.9 Hz, H-1″)*, 4.11 (t, 1H, J2″-1″~2″-3″ = 9.3 Hz, H-2″), 3.97 (br d, 1H, J6″a-6″b = 12.1 Hz, H-6″a)*, 3.81 (dd, 1H, J6″b-6″a = 12.1 Hz, J6″b-5″ = 5.3 Hz, H-6″a), 3.68 (t, 1H, J4″-3″~4″-5″ = 9.2 Hz, H-4″), 3.55–3.48 (m, 2H, H-3″ and H-5″). 13C NMR (MeOD) δ (ppm) 184.2 (C-4), 166.0 (C-2), 164.8 (C-7), 162.8 (C-5), 158.2 (C-8a), 133.1 (C-3′ and C-5′), 132.8 (C-1′), 130.2 (C-4′), 128.1, 127.8 (C-2′ and C-6′)*, 105.8 (C-3), 105.1 (C-8), 104.6 (C-4a), 99.6 (C-6), 82.9 (C-5″), 80.2 (C-3″), 75.3 (C-1″), 72.8 (C-2″), 72.3, 71.5 (C-4″)*, 63.1, 62.7 (C-6″)*. *Two peaks were observed due to the presence of rotamers. HRMS-ESI (m/z): [M + H]+ calcd for C21H21O9 417.1180, found 417.1174.
8-(β-d-Glucopyranosyl)-5,7,4′-trihydroxyflavone (17). Purified by column chromatography (DCM/MeOH 1:0 to 5:1). Isolated yield over two steps: 7%; Rf = 0.43 (EtOAc/EtOH 6:1). 1H NMR (MeOD) δ (ppm) 7.93 (d, 2H, Jortho = 8.6 Hz, H-2′, H-6′), 6.91 (d, 2H, Jortho = 8.8 Hz, H-3′, H-5′), 6.49 (s, 1H, H-3), 6.14 (s, 1H, H-6), 5.05 (d, 1H, J1″-2″ = 9.7 Hz, H-1″), 4.17–4.10 (m, 1H, H-2″), 3.94 (d, 1H, J6″a-6″b = 12.0 Hz, H-6″a), 3.79 (dd, 1H, J6″b-6″a = 12.1 Hz, J6″b-5″ = 5.5 Hz, H-6″b), 3.65 (t, 1H, J4″-3″ = J 4″-5″ = 9.2 Hz, H-4″), 3.56–3.48 (m, 2H, H-3″, H-5″). 13C NMR (MeOD) δ (ppm) 182.1 (C-4), 168.2 (C-4′), 164.4 (C-7), 163.3 (C-2), 161.2 (C-5), 160.8 (C-8a), 129.8 (C-2′, C-6′), 124.6 (C-1′), 117.3 (C-3′, C-5′), 106.2 (C-4a), 104.5 (C-8), 102.8 (C-3), 99.3 (C-6), 82.8 (C-2″), 80.7 (C-5″), 78.5 (C-3″), 74.4 (C-1″), 72.5 (C-4″), 61.4 (C-6″). HRMS-ESI (m/z): [M + H]+ calcd for C21H21O10 433.1129, found 433.1120.
4′-Fluoro-8-(β-d-glucopyranosyl)-5,7-dihydroxyflavone (18). Purified by preparative HPLC. Reaction yield over two steps: 45%; LCMS: RT= 0.71 min, m/z = 433.00 [M - H]- (low pH method); colorless oil. 1H NMR (MeOD) δ (ppm) 8.12 (dd, 2H, Jortho = 8.20 Hz, JH-F = 5.9 Hz, H-2′, H-6′), 7.27 (t, 2H, Jortho~H-F = 8.7 Hz, H-3′, H-5′), 6.52 (s, 1H, H-3), 6.02 (s, 1H, H-6), 5.04 (d, 1H, J1″-2″ = 9.3 Hz, H-1″), 4.11 (t, 1H, J2″-1″ = J 2″-3″ = 9.2 Hz, H-2″), 3.89 (dd, 1H, J6″a-6″b = 12.3 Hz, J6″a-5″ = 1.8 Hz, H-6″a), 3.80 (dd, 1H, J6″b-6″a = 12.1 Hz, J6″b-5″ = 4.8 Hz, H-6″b), 3.65 (t, 1H, J4″-3″ = J 4″-5″ = 9.3 Hz, H-4″), 3.51 (t, 1H, J3″-2″ = J 3″-4″ = 9.0 Hz, H-3″), 3.48–3.44 (m, 1H, H-5″). 13C NMR (MeOD) δ (ppm) 182.5 (C-4), 167.5 (C-2), 166.0 (d, JC-F = 251.2 Hz, C-4′), 163.3 (C-7), 161.9 (C-5), 159.1 (C-8a), 130.3 (d, JC-F = 8.8 Hz, C-2′, C-6′), 129.9 (d, JC-F = 3.2 Hz, C-1′), 117.0 (d, JC-F = 22.5 Hz, C-3′, C-5′), 105.6 (C-4a), 104.8 (C-3), 104.4 (C-6), 104.1 (C-8), 82.6 (C-5″), 80.9 (C-3″), 76.0 (C-1″), 73.8 (C-2″), 72.1 (C-4″), 62.8 (C-6″). HRMS-ESI (m/z): [M + H]+ calcd for C21H20FO9 435.1086, found 435.1083.
8-(β-d-Glucopyranosyl)-5,7-dihydroxy-4′-(morpholin-4-yl)flavone (19). Purified by preparative HPLC. Reaction yield over two steps: 74%; LCMS: RT = 0.57 min, m/z = 500.0 [M − H]− (high pH method); orange solid; m.p. = 210.5–211.4 °C; = + 10 (c 0.5 MeOH); 1H NMR (MeOD) δ (ppm) 7.94, 7.84 (d, 2H, Jortho = 8.3 Hz, H-2′ and H-6′)*, 7.01 (d, 2H, Jortho = 8.6 Hz, H-3′ and H-5′), 6.52 (s, 1H, H-3), 6.26 (s, 1H, H-6), 5.05, 4.99 (d, 1H, J1″-2″ = 9.9 Hz, H-1″)*, 4.14 (t, 1H, J2″-1″ = J 2″-3″ = 9.5 Hz, H-2″), 3.98–3.79 (m, 6H, H-6″a, H-6″b and NCH2CH2O), 3.70 (t, 1H, J4″-3″ = J 4″-5″ = 9.6 Hz, H-4″), 3.57–3.53 (m, 1H, H-3″), 3.49–3.46 (m, 1H, H-5″), 3.32–3.20 (NCH2CH2O, superimposed with the MeOD peak). 13C NMR (MeOD) δ (ppm) 184.0 (C-4), 166.5 (C-2), 164.4 (C-7), 162.6 (C-5), 158.0 (C-8a), 155.1 (C-4′), 129.5, 129.1 (C-2′ and C-6′)*, 122.0 (C-1′), 115.3 (C-3′ and C-5′), 105.8 (C-4a), 105.2 (C-8), 103.0 (C-3), 99.4 (C-6), 82.8 (C-5″), 80.3 (C-3″), 75.3 (C-1″), 72.9 (C-2″), 72.3 (C-4″), 67.7 (NCH2CH2O), 63.1 (C-6″), 40.4 (NCH2CH2O). *Peaks were observed due to the presence of rotamers. HRMS-ESI (m/z): [M + H]+ calcd for C25H28NO10 502.1708, found 502.1695.
8-(β-d-Glucopyranosyl)-5,7-dihydroxy-4′-(pyrrolidin-1-yl)flavone (20). Purified by preparative HPLC. Reaction yield over two steps: 80%; LCMS: RT = 0.86 min, m/z = 486.00 [M + H]+ (low pH method); orange oil. 1H NMR (MeOD) δ (ppm) 7.93 (d, 2H, Jortho = 8.2 Hz, H-2′ and H-6′)*, 6.67 (d, 2H, Jortho = 8.3 Hz, H-3′ and H-5′), 6.50 (s, 1H, H-3), 6.25 (s, 1H, H-6), 4.99 (d, 1H, J1″-2″ = 9.9 Hz, H-1″)*, 4.16 (t, 1H, J2″-1″ =J2″-3″ = 9.4 Hz, H-2″), 3.98 (d, 1H, J6-a″- 6-b″ = 11.8 Hz, H-6″a)*, 3.80 (dd, 1H, J6-b″-6-a″ = 11.9 Hz, J6-b″,5″ = 5.8 Hz, H-6″b), 3.70 (t, 1H, J4″-3″ = J 4″-5″ = 9.2 Hz, H-4″), 3.56–3.46 (m, 2H, H-3″, H-5″), 3.38–3.35 (m, 4H, NCH2CH2), 2.07 (s, 4H, NCH2CH2). 13C NMR (MeOD) δ (ppm) 181.6 (C-4), 167.6 (C-2), 165.5 (C-7), 162.6 (C-5), 160.4 (C-8a), 156.4 (C-4′), 129.7 (C-2′ and C-6′), 121.7 (C-1′), 112.8 (C-3′ and C-5′), 105.0 (C-4a), 104.5 (C-8), 101.5 (C-3), 97.9 (C-6), 82.9 (C-5″), 80.3 (C-3″), 75.3 (C-1″), 72.9 (C-2″), 72.4 (C-4″), 63.2 (C-6″), 49.1 (NCH2CH2, overlapped with the MeOD peak), 26.4 (NCH2CH2). *Peaks were observed due to the presence of rotamers. HRMS-ESI (m/z): [M + H]+ calcd for C25H28NO9 486.1759, found 486.1743.
2-(Furan-2-yl)-8-(β-d-glucopyranosyl)-5,7-dihydroxy-4H-chromen-4-one (21). Purified by preparative HPLC. Reaction yield over two steps: 56%; LCMS: RT = 0.62 min, m/z = 404.80 [M − H]− (low pH method); yellowish oil. 1H NMR (MeOD) δ (ppm) 7.82 (d, 1H, J2′-3′ = 3.5 Hz, H-2′), 7.33 (br s, 1H, H-4′), 6.72 (dd, 1H, J3′-4′ = 1.8 Hz, H-3′), 6.53 (s, 1H, H-3), 6.28 (s, 1H, H-6), 4.96 (d, 1H, J1″-2″ = 9.4 Hz, H-1″), 4.16 (t, 1H, J2″-1″ = J2″-3″ = 9.4 Hz, H-2″), 3.89 (d, 1H, J6-a″,6-b″ = 11.6 Hz, H-6″a), 3.73 (dd, 1H, J6-b″ = J 5″ = 5.1 Hz, H-6″b), 3.64 (t, 1H, J4″-3″ = J 4″-5″ = 9.4 Hz, H-4″), 3.53 (t, 1H, J4″-3″~4″-5″ = 9.2 Hz, H-3″), 3.46–3.41 (m, 1H, H-5″). 13C NMR (MeOD) δ 183.7 (C-4), 167.2 (C-2), 164.9 (C-7), 162.9 (C-5), 157.3 (C-8a), 148.0 (C-2′), 147.3 (C-1′), 115.6 (C-4′), 113.9 (C-3′), 107.2 (C-4a), 106.0 (C-8), 103.5 (C-3), 99.6 (C-6), 82.6 (C-5″), 80.1 (C-3″), 75.0 (C-1″), 72.9 (C-2″), 72.3 (C-4″), 63.0 (C-6″). HRMS-ESI (m/z): [M + H]+ calcd for C19H19NO10 407.0973, found 407.0965.
8-(β-d-Glucopyranosyl)-5,7-dihydroxy-4H-chromen-4-one (22). Compound 42 (0.195 g, 0.221 mmol, 1 eq.) was dissolved in anhydrous DCM (5 mL). The mixture was stirred at −78 °C, and BCl3 (1M solution in DCM, 2.21 mL, 2.21 mmol, 10 eq.) was added in a dropwise manner over 5 min. The reaction was complete after 1 h, as detected by LCMS, and was quenched with a 1:1 mixture of DCM/MeOH (40 mL). The mixture was stirred for approximately 20 min at room temperature. The solvent was evaporated under vacuum and the residue purified by column chromatography (DCM-MeOH 1:0 to 4:1). Compound 22 was obtained as a white solid; m.p. = 192.5–193.0 °C; = + 16 º (c 0.5 MeOH). Reaction yield: 45%; LCMS: RT = 0.41 min, m/z = 338.80 [M - H]- (low pH method). 1H NMR (MeOD) δ (ppm) 8.04 (d, 1H, Jcis = 5.8 Hz, H-2), 6.28 (s, 1H, H-6), 6.24 (d, 1H, Jcis = 5.9 Hz, H-3), 4.91 (d, 1H, J1″-2″ = 9.9 Hz, H-1″), 4.07 (t, 1H, J2″-1″~2″-3″ = 9.3 Hz, H-2″), 3.88 (dd, 1H, J6-a″~6-b″ = 12.1 Hz, J6-a″~5″ = 2.1 Hz, H-6″a), 3.71 (dd, 1H, J6-b″~6-a″ = 12.0 Hz, J6-b″~5″ = 5.4 Hz, H-6″b), 3.50–3.41 (m, 3H, H-3″, H-4″, H-5″). 13C NMR (MeOD) δ 183.7 (C-4), 165.0 (C-7), 163.0 (C-5), 158.0 (C-8a and C-2), 111.5 (C-3), 106.8 (C-4a), 104.9 (C-8), 100.5 (C-6), 82.6 (C-5″), 80.1 (C-3″), 75.4 (C-1″), 72.8 (C-2″), 71.8 (C-4″), 62.9 (C-6″). HRMS-ESI (m/z): [M + H]+ calcd for C15H17NO9 341.0867, found 341.0865.
LogD7.4 determination. The in-silico prediction tool ALOGPS [
4] was used to estimate the octanol-water partition coefficients (log
P) of the compounds. Depending on these values, the compounds were classified either as hydrophilic (log
P below zero), moderately lipophilic (log
P between zero and one), or lipophilic (log
P above one) compounds. For each category, two different ratios (volume of octan-1-ol to volume of buffer) were defined as experimental parameters (
Table 2).
Equal amounts of phosphate buffer (0.1 M, pH 7.4) and octan-1-ol were mixed and shaken vigorously for 5 min to saturate the phases. The mixture was left until separation of the two phases, and the buffer was retrieved. Stock solutions of the test compounds were diluted with buffer to a concentration of 1 μM. For each compound, three determinations per octan-1-ol:buffer ratio were performed in different wells of a 96-well plate. The respective volumes of buffer-containing analyte (1 μM) were pipetted to the wells and covered by saturated octan-1-ol according to the chosen volume ratio. The plate was sealed with aluminum foil, shaken (1350 rpm, 25 °C, 2 h) on a Heidolph Titramax 1,000 plate-shaker (Heidolph Instruments GmbH & Co. KG, Schwabach, Germany) and centrifuged (2,000 rpm, 25 °C, 5 min, 5804 R Eppendorf centrifuge, Hamburg, Germany). The aqueous phase was transferred to a 96-well plate for analysis by liquid chromatography-mass spectrometry (LCMS, see below). Log
P coefficients were calculated from the octan-1-ol:buffer ratio (o:b), the initial concentration of the analyte in buffer (1 μM), and the concentration of the analyte in buffer (
cB) according to the following equation:
Results are presented as the mean ± SD of three independent experiments. If the mean of two independent experiments obtained for a given compound did not differ by more than 0.1 units, the results were accepted.
Parallel artificial membrane permeability assay (PAMPA). Effective permeability (log
Pe) was determined in a 96-well format with PAMPA [
34]. For each compound, measurements were performed at pH 7.4 in quadruplicates. Four wells of a deep well-plate were filled with 650 μL of PRISMA HT universal buffer, adjusted to pH 7.4 by adding the requested amount of NaOH (0.5 M). Samples (150 μL) were withdrawn from each well to determine the blank spectra by UV/Vis-spectroscopy (190 to 500 nm, SpectraMax 190, Molecular Devices, Silicon Valley, CA, USA). Then the analyte, dissolved in DMSO (10 mM), was added to the remaining buffer to yield 50 μM solutions. To exclude precipitation, the optical density (OD) was measured at 650 nm, and solutions exceeding OD 0.01 were filtrated. Afterwards, samples (150 μL) were withdrawn to determine the reference spectra. Further 200 μL were transferred to each well of the donor plate of the PAMPA sandwich (pIon, P/N 110 163). The filter membranes at the bottom of the acceptor plate were infused with 5 μL of GIT-0 Lipid Solution and 200 μL of Acceptor Sink Buffer were filled into each acceptor well. The sandwich was assembled, placed in the GutBox
TM, and left undisturbed for 16 h. Then, it was disassembled and samples (150 μL) were transferred from each donor and acceptor well to UV-plates for determination of the UV/Vis spectra. Effective permeability (log
Pe) was calculated from the compound flux deduced from the spectra, the filter area, and the initial sample concentration in the donor well with the aid of the PAMPA Explorer Software (pIon, version 3.5).
LC-MS measurements. Analyses were performed using a 1100/1200 Series HPLC System coupled to a 6410 Triple Quadrupole mass detector (Agilent Technologies, Inc., Santa Clara, CA, USA) equipped with electrospray ionization. The system was controlled with the Agilent MassHunter Workstation Data Acquisition software (version B.01.04). The column used was an AtlantisR T3 C18 column (2.1 × 50 mm) with a 3 μm-particle size (Waters Corp., Milford, MA, USA). The mobile phase consisted of eluent A: 10 mM ammonium acetate, pH 5.0 in 95:5, H2O:MeCN; and eluent B: MeCN containing 0.1% formic acid. The flow rate was maintained at 0.6 mL/min. The gradient was ramped from 95% A/5% B to 5% A/95% B over 1 min, and then held at 5% A/95% B for 0.1 min. The system was then brought back to 95% A/5% B, resulting in a total duration of 4 min. MS parameters such as fragmentor voltage, collision energy, and polarity were optimized individually for each drug, and the molecular ion was followed for each compound in the multiple reaction monitoring mode. The concentrations of the analytes were quantified by the Agilent Mass Hunter Quantitative Analysis software (version B.01.04).
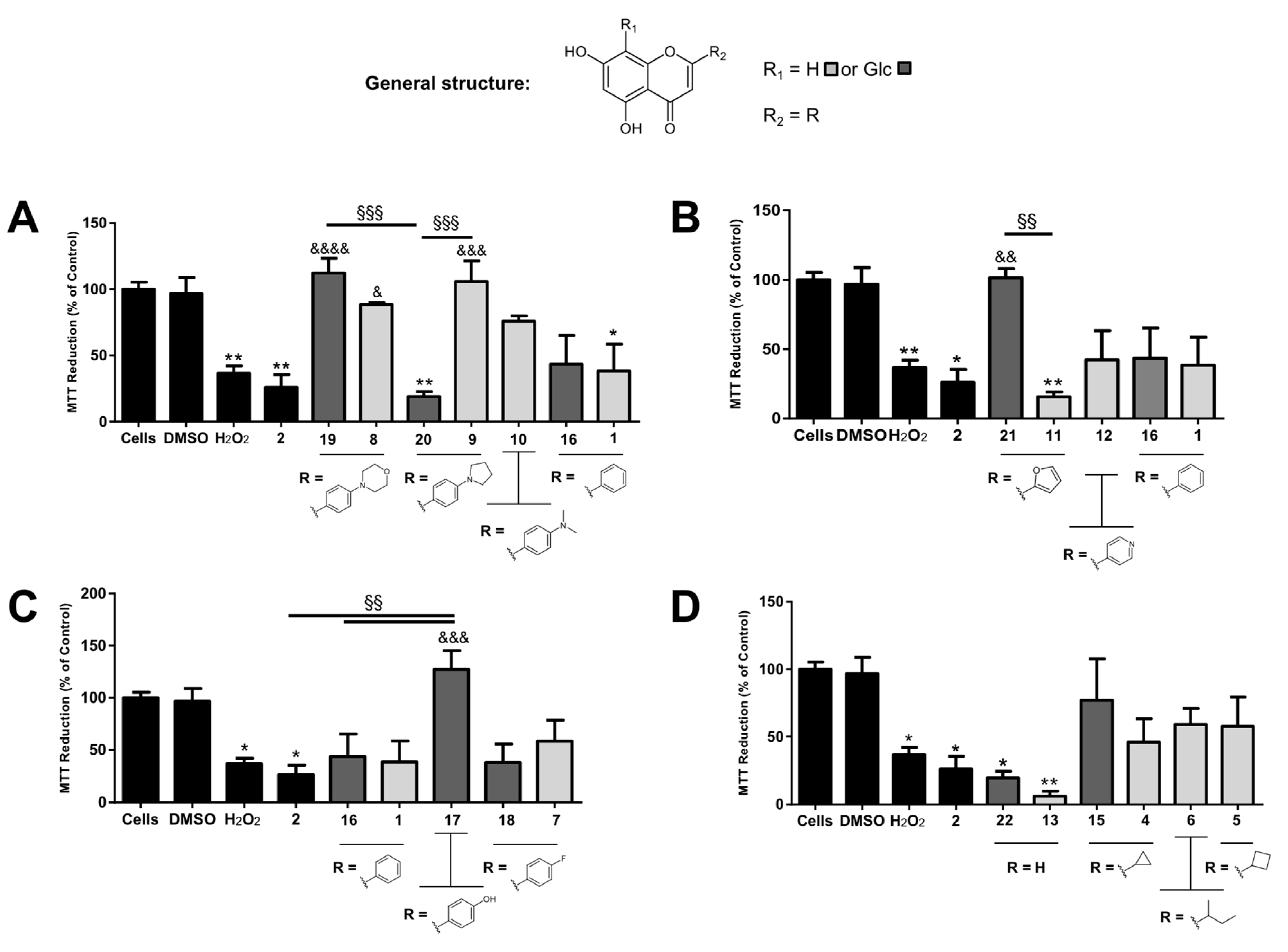
Neuroprotective assays in human neuroblastoma (SH-SY5Y) cells. SH-SY5Y cells were grown in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Biochrom GmbH) and 1% Penicillin-Streptomycin (Gibco, Life Technologies) in a humidified incubator at 37 °C, 5% CO
2. For the neuroprotective activity assay, undifferentiated SGSY-5Y cells were plated onto 96-well flat-bottomed microtiter plates at a density of 1 × 10
4 cells/well in DMEM supplemented with 2% FBS and preincubated for 24 h at 37 °C, 5% CO
2. Compounds (stored as 10 mM solutions in DMSO at −20 °C) were then added to achieve a final concentration of 50 μM and, after 30 min, cells were incubated in the presence or absence of 20 μM Aβ protein fragment 1-42 (Sigma-Aldrich, dissolved in DMSO and stored in 2 mM aliquots at −20 °C) or 100 μM of H
2O
2 (Sigma-Aldrich, dissolved to 10 mM in 0.9% NaCl aqueous solution immediately prior to the assay) overnight at 37 °C, 5% CO
2. The final DMSO percentage was 0.5% for compounds in the presence of H
2O
2, 1% for cells incubated only with Aβ, or 1.5% for cells incubated with compounds and Aβ, thus, controls for each DMSO percentage were also run. The following morning, 20 μL of a 5 mg/mL solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich) in PBS (Gibco, Life Technologies) was added to each well and the plates were further incubated for 4 h at 37 °C, followed by the addition of DMSO (200 μL) to each well in order to dissolve the resulting formazan crystals. After 2 h incubating at 37 °C, the optical density (OD) at 540 nm (with a 620 nm reference filter) was measured in an Amersham Biosciences Biotrak II Plate Reader. The percentage of MTT reduction was determined according to Equation (1). All experiments were performed in triplicate and results are presented as means ± standard error. Differences between experimental conditions were compared for statistical significance by one-way ANOVA followed by a Tukey’s post-test, an analysis carried out using GraphPad Prism Software (LA Jolla, CA, USA). Differences were considered significant when
P < 0.05. In order to exclude direct MTT reduction, compounds were also tested in (a) the absence of cells and (b) in the absence of both cells and culture medium, using the same experimental conditions described above.
Cell viability assay in human epithelial colorectal adenocarcinoma (Caco-2) and human liver hepatocellular (HepG2) cells. Caco-2 and HepG2 Cells were cultured in DMEM-Dulbecco’s modified Eagle’s medium (Sigma-Aldrich) supplemented with 10% (v/v) inactivated fetal bovine serum (PAA Laboratories GmbH), 2 mM l-glutamine (Sigma-Aldrich), 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich), in a humidified incubator at 37 °C with a 5% CO2 atmosphere. Cells (3 × 104 cells/well) were seeded into 96-well plates and incubated at 37 °C in 5% CO2 atmosphere. After 24 h, compounds were added at different concentrations (0.1–100 μM) and incubated in the same conditions. DMSO controls were performed to evaluate a possible solvent cytotoxicity, while pure DMSO was used as a cytotoxic drug. After the established incubation time with compounds, MTT (5 mg/mL) in PBS (Sigma-Aldrich) was added (10 μL) to each well. After 3 h incubation at 37 °C, in 5% CO2, the supernatant was removed. The formazan crystals were solubilized using ethanol/DMSO (1:1) (100 μL), and the absorbance values were determined at 570 nm on the microplate reader Victor3 from PerkinElmer Life Sciences.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}