Generation of Stable cisPt Resistant Lung Adenocarcinoma Cells

 , ,
, ,  ,
,
Abstract
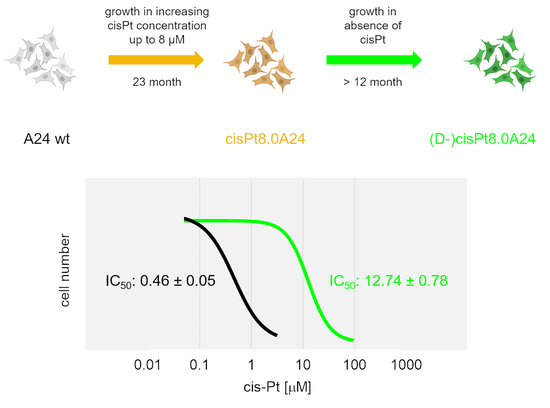
:
1. Introduction
2. Results
2.1. Immunocytochemical Verification of Lung Adenocarcinoma Properties of A24 wt Cells
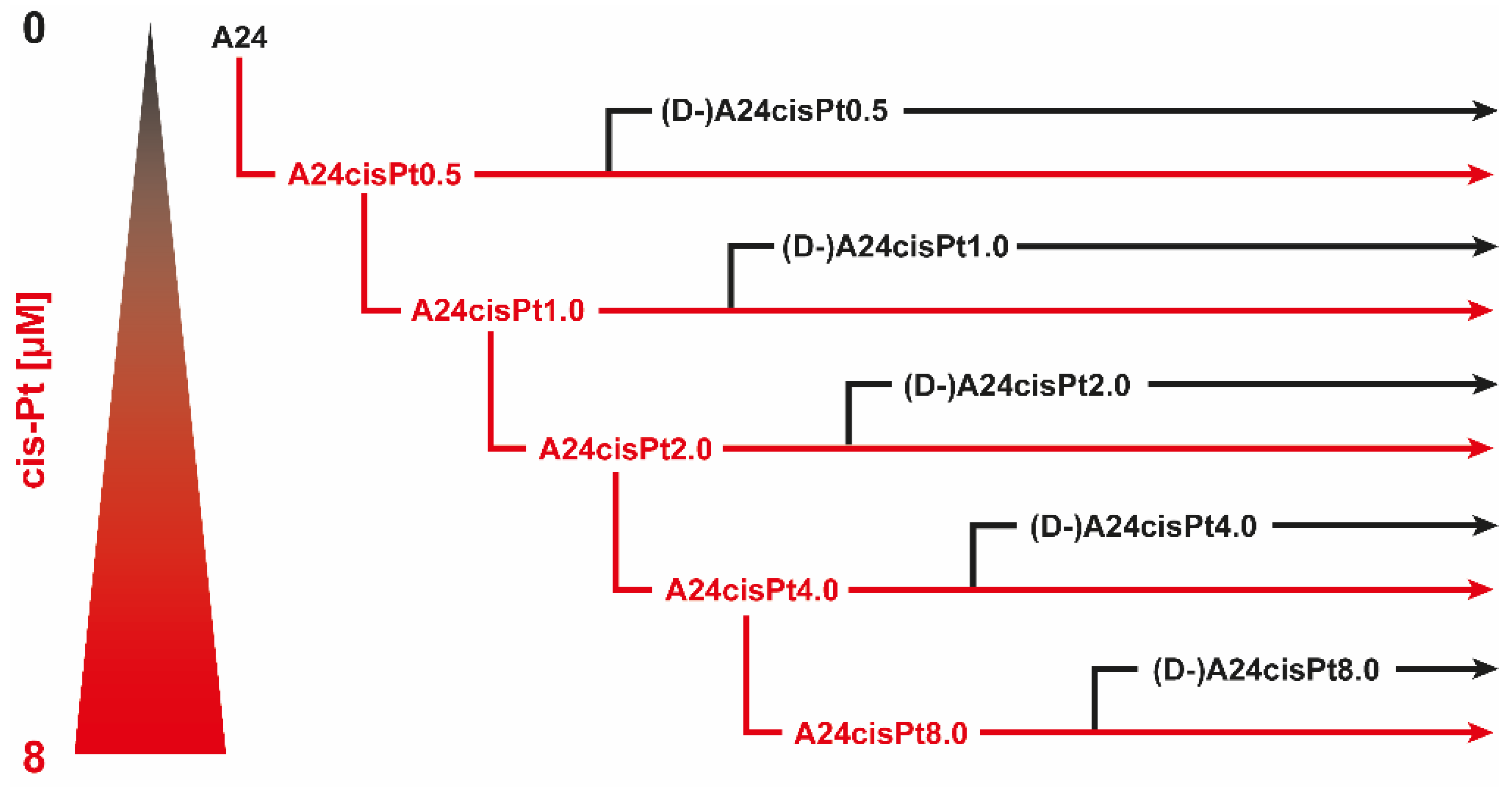
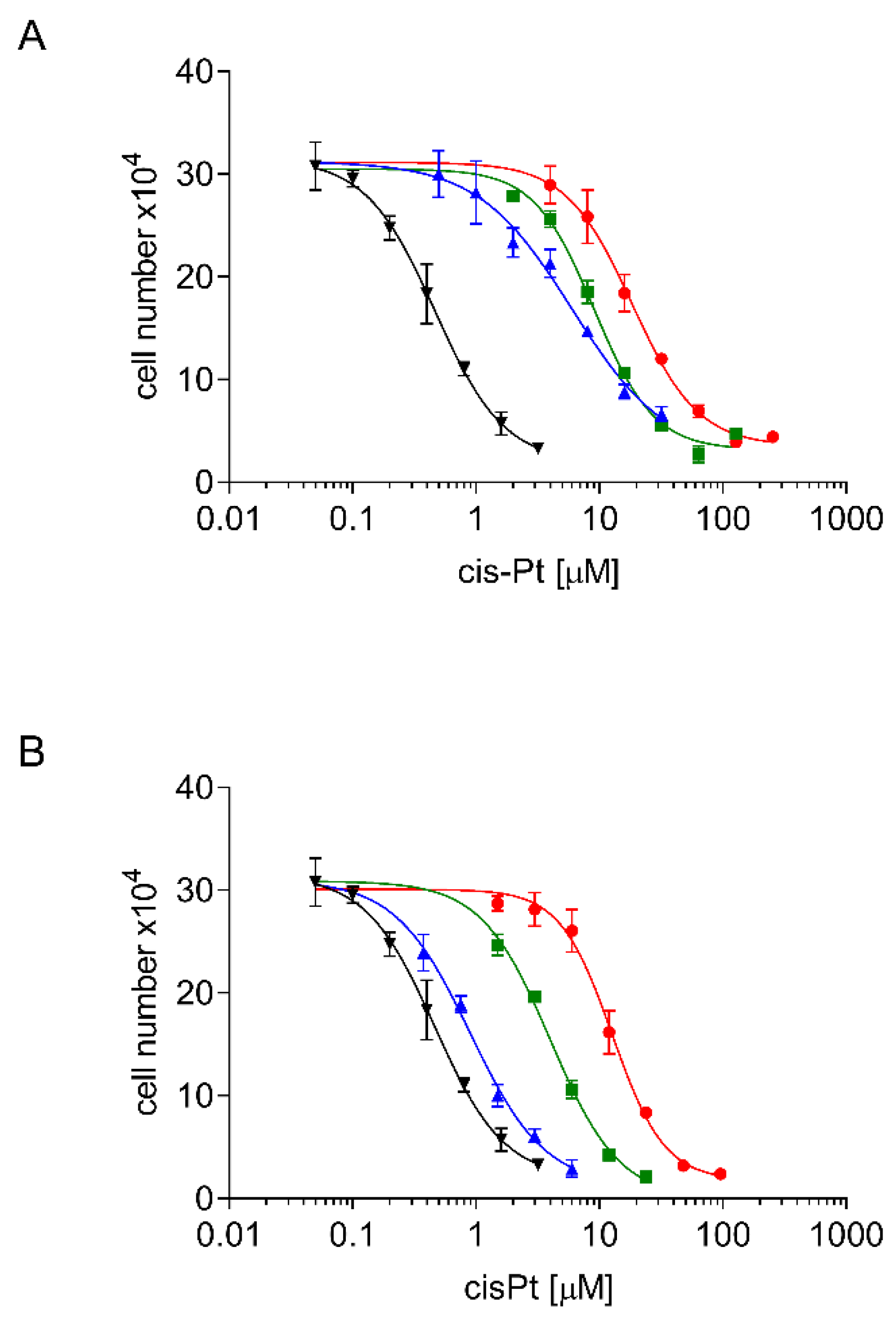
2.2. Development, Levels, and Stability of Graded cisPt Resistance in A24cisPt and (D-)A24cisPt Sublines
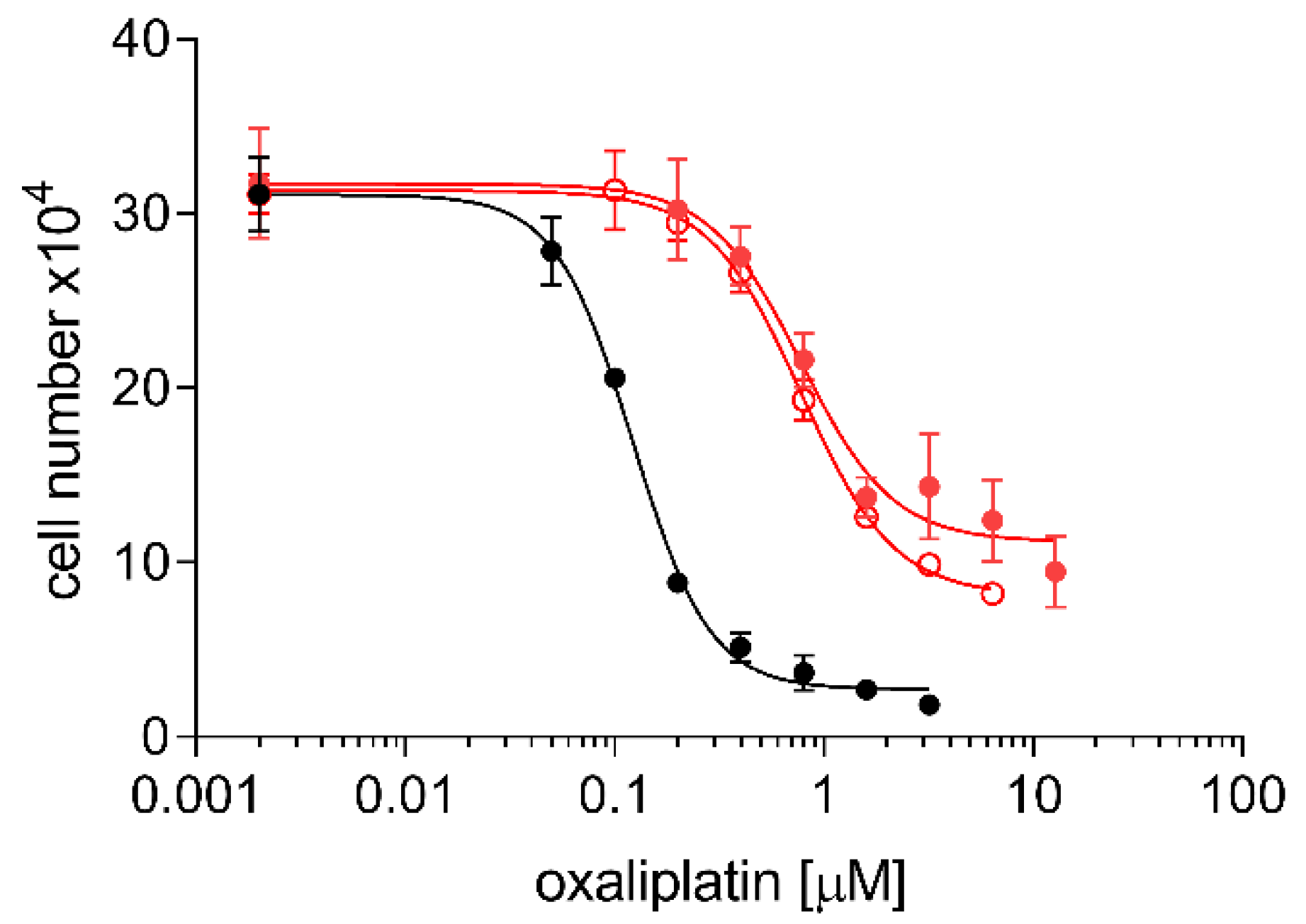
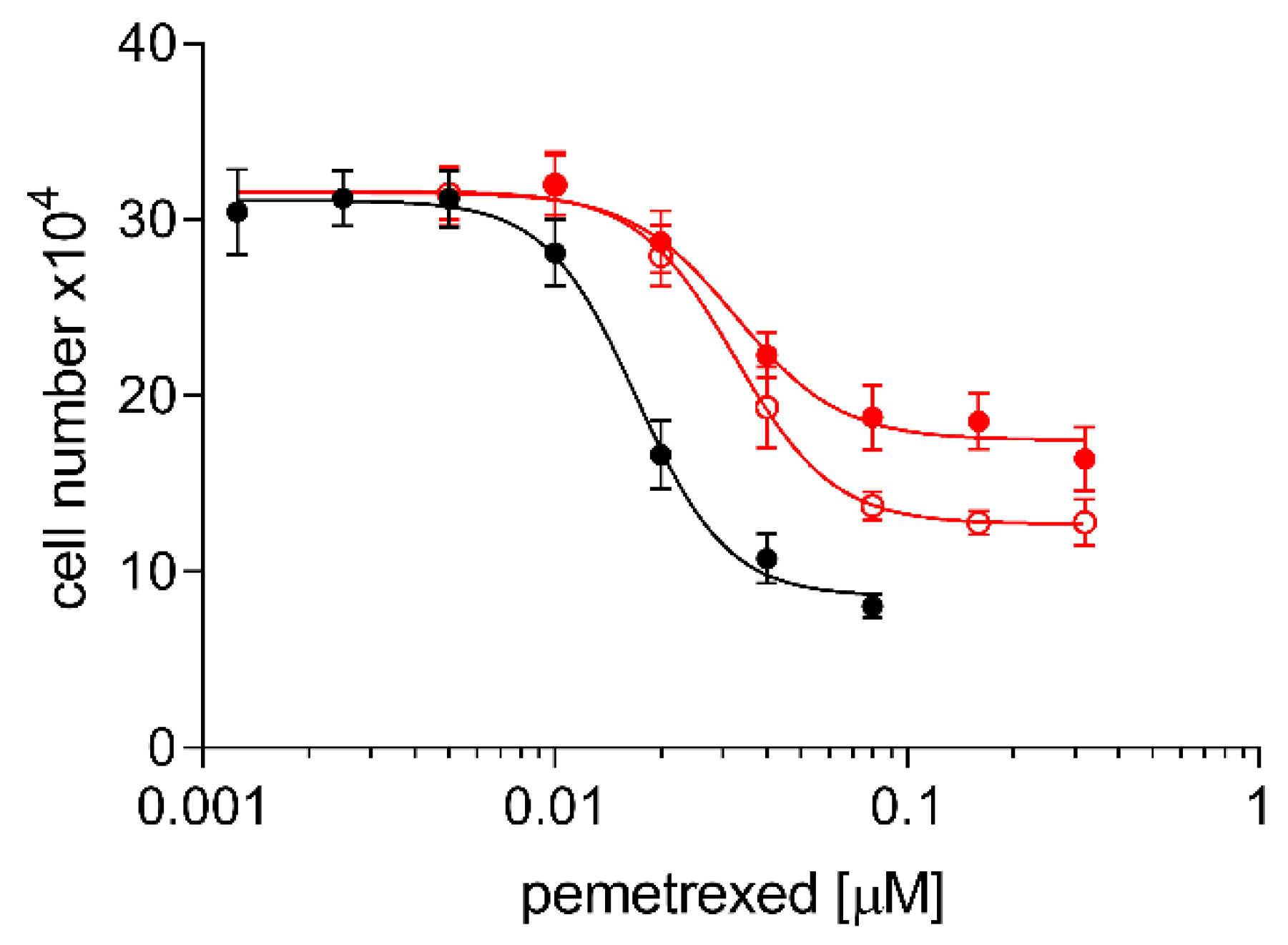
2.3. Cross Resistance of A24cisPt and (D-)A24cisPt Sublines to Oxaliplatin and Pemetrexed
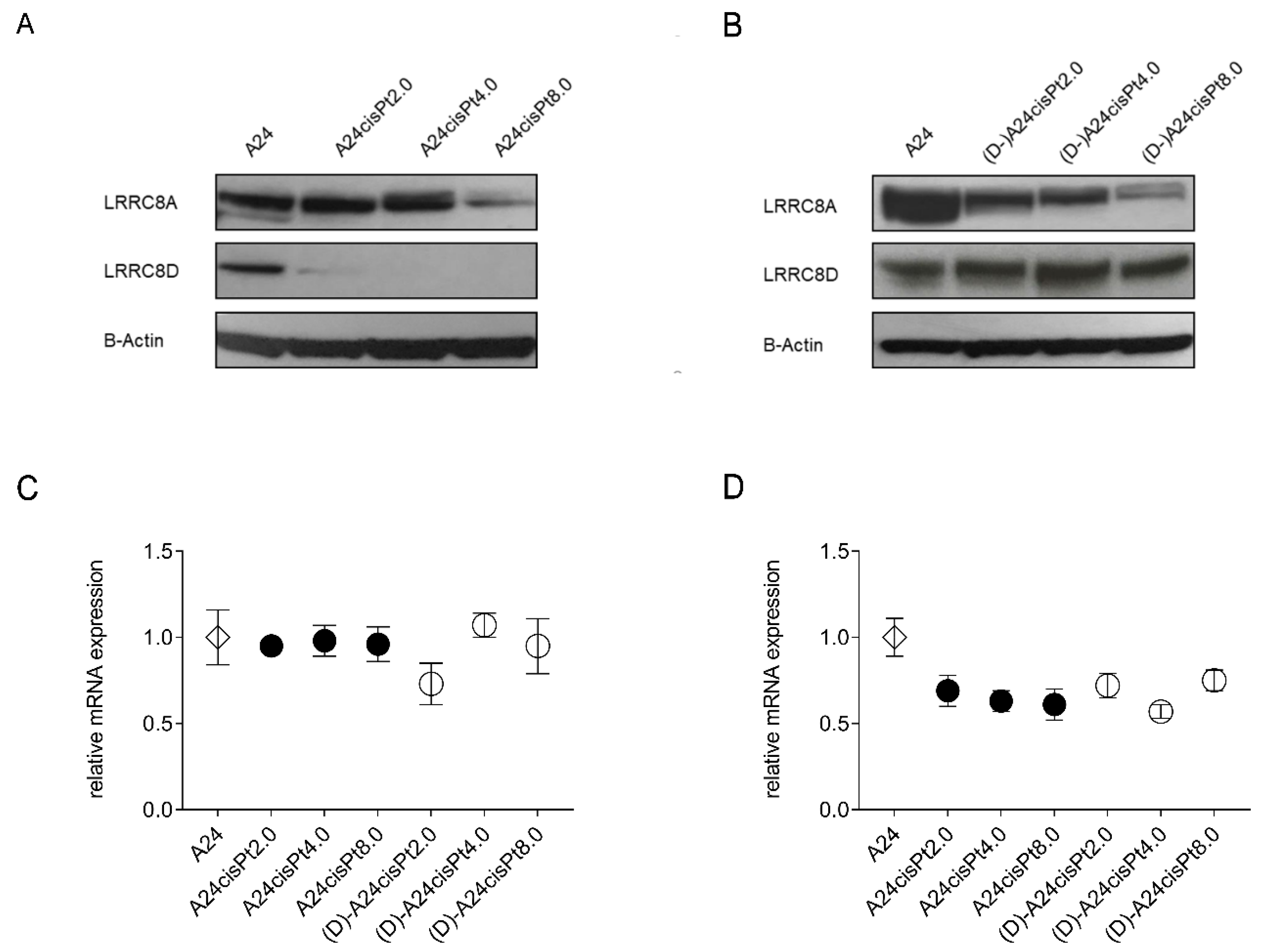
2.4. Expression of VRAC Subunits in A24 wt, A24cisPt, and (D-)A24cisPt Cells
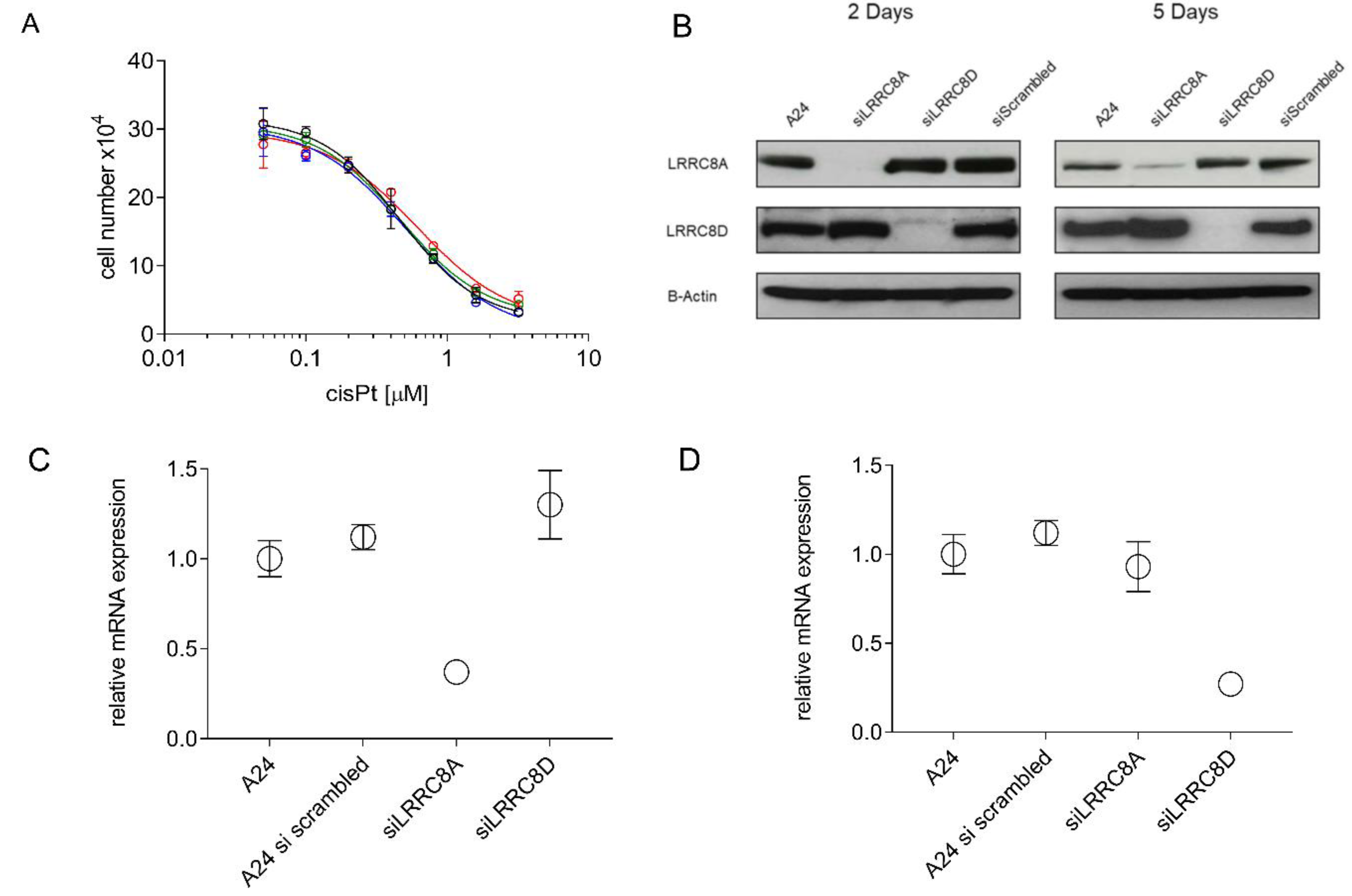
2.5. CisPt Response of siLRRC8A or siLRRC8D Transfected A24 wt Cells
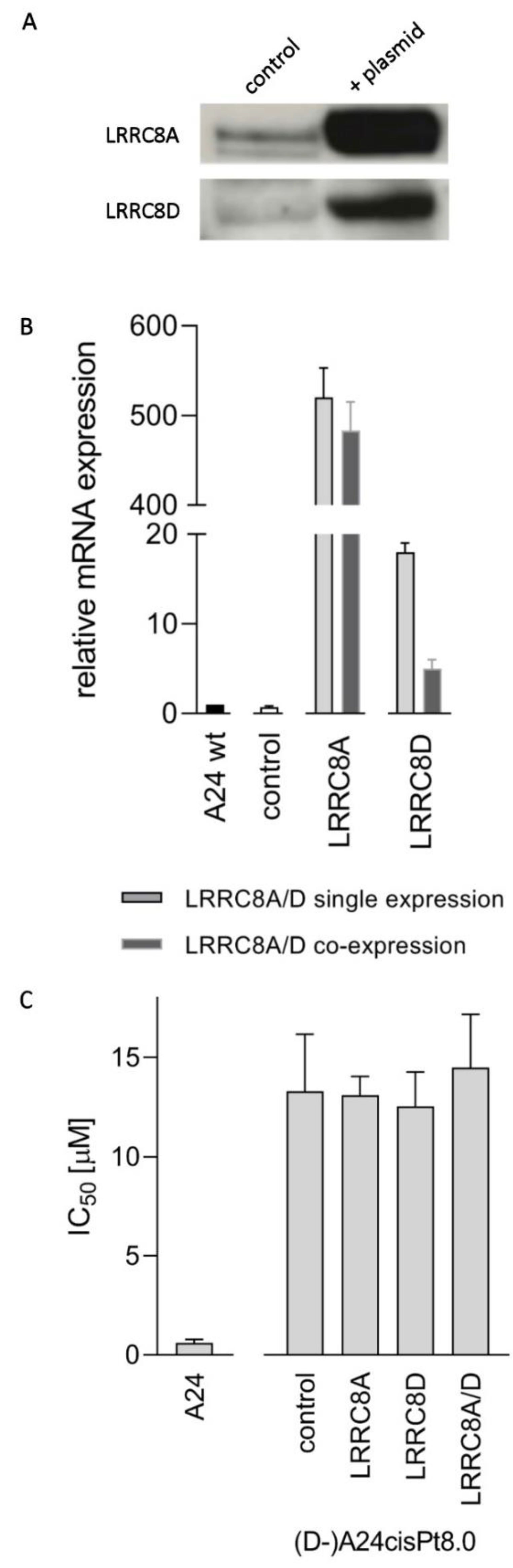
2.6. CisPt Response in Transfected (D-)A24cisPt8.0 Cells Overexpressing LRRC8A or LRRC8D
3. Discussion
4. Materials and Methods
4.1. Platinum Derivatives and Pemetrexed
4.2. Metastatic Lung Adenocarcinoma Cells and Cell Cultivation
4.3. Immunofluorescence
4.4. Induction, Maintenance, and De-Induction of cisPt Resistance
4.5. IC50 Determination
4.6. Western Blotting
4.7. siRNA Cell Assay in A24 wt Cells
4.8. Overexpression Cell Assay in (D-)A24cisPt8.0 Cells
4.9. Reverse Transcription Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
- LRRC8A forward (5′-GGTTTGCCAAGTACTTCCCCTAC-3′),
- LRRC8A reverse (5′-CTTCAGCAGGATAGACACAAAGTG-3′),
- LRRC8D forward (5′-CATGCAACTTACCAAAGATCAGGTG-3′),
- LRRC8D reverse (5′-CTGCTTCCATCTTTGGGATGTTG-3′),
- β-Actin forward (5′-GATGGTGGGCATGGGTC-3′) and
- β-Actin reverse (5′-GATTTTCTCCATGTCGTCCCAG -3′).
4.10. Statistics
4.11. Ethical Statements
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cisPt | Cisplatin |
| wt | Wild-type |
| A24 | A240286S |
| NSCLC | Non-small cell lung cancer |
| SCLC | Small cell lung cancer |
| TTF-1 | Thyroid transcription factor 1 |
| LRRC8A | Leucine-rich repeat-containing protein 8A |
| LRRC8D | Leucine-rich repeat-containing protein 8D |
| p63 | Tumor protein p63 |
| VEGF | Vascular endothelial growth factor |
| VRACs | Volume-regulated anion channels |
References
- Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Yang, Y.; Yang, Y.; Zhang, Y.; Yue, Z.; Pan, Z.; Ren, X. Ginsenoside Rg3 attenuates cisplatin resistance in lung cancer by downregulating PD-L1 and resuming immune. Biomed. Pharmacother. 2017, 96, 378–383. [Google Scholar] [CrossRef]
- Tian, Y.; Sun, C.; Zhang, L.; Pan, Y. Clinical significance of miRNA—106a in non-small cell lung cancer patients who received cisplatin combined with gemcitabine chemotherapy. Cancer Biol. Med. 2018, 15, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L.R. Preclinical perspectives on platinum resistance. Drugs 2000, 59 (Suppl. 4), 1–8. [Google Scholar] [CrossRef]
- Yoshida, M.; Khokhar, A.R.; Siddik, Z.H. Biochemical pharmacology of homologous alicyclic mixed amine platinum(II) complexes in sensitive and resistant tumor cell lines. Cancer Res. 1994, 54, 3468–3473. [Google Scholar]
- Kalayda, G.V.; Wagner, C.H.; Jaehde, U. Relevance of copper transporter 1 for cisplatin resistance in human ovarian carcinoma cells. J. Inorg. Biochem. 2012, 116, 1–10. [Google Scholar] [CrossRef]
- Samimi, G.; Safaei, R.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Goodman, M.; Howell, S.B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [Green Version]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- Voets, T.; Nilius, B.; Vennekens, R. VRACs swallow platinum drugs. EMBO J. 2015, 34, 2985–2987. [Google Scholar] [CrossRef] [Green Version]
- Rubino, S.; Bach, M.D.; Schober, A.L.; Lambert, I.H.; Mongin, A.A. Downregulation of Leucine-Rich Repeat-Containing 8A Limits Proliferation and Increases Sensitivity of Glioblastoma to Temozolomide and Carmustine. Front. Oncol. 2018, 8, 142. [Google Scholar] [CrossRef]
- Granzow, C. Method for Creating a Database for the Preliminary Estimation of the Effectiveness of Active Substances in Tumor Therapy. European Patent Office EP3013972B1, 22 August 2018. [Google Scholar]
- Ruprecht, N.O.; Hungerbuhler, M.; Granzow, C.; Kempf, C.M.; Heverhagen, J.T. Cisplatin response of NSCLC cells irresponsive to knocking down VRAC subunits LRRC8A or LRRC8D. In Proceedings of the AACR Annual Meeting, Chicago, IL, USA, 14–18 April 2018; p. 2844. [Google Scholar]
- Carney, J.M.; Kraynie, A.M.; Roggli, V.L. Immunostaining in lung cancer for the clinician. Commonly used markers for differentiating primary and metastatic pulmonary tumors. Ann. Am. Thorac. Soc. 2015, 12, 429–435. [Google Scholar] [CrossRef]
- Voss, F.K.; Ullrich, F.; Munch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desoize, B.; Berthiot, G.; Manot, L.; Coninx, P.; Dumont, P. Evaluation of a prediction model of cisplatin dose based on total platinum plasma concentration. Eur. J. Cancer 1996, 32, 1734–1738. [Google Scholar] [CrossRef]
- Lai, G.M.; Ozols, R.F.; Smyth, J.F.; Young, R.C.; Hamilton, T.C. Enhanced DNA repair and resistance to cisplatin in human ovarian cancer. Biochem. Pharmacol. 1988, 37, 4597–4600. [Google Scholar] [CrossRef]
- Fujii, R.; Mutoh, M.; Sumizawa, T.; Chen, Z.S.; Yoshimura, A.; Akiyama, S. Adenosine triphosphate-dependent transport of leukotriene C4 by membrane vesicles prepared from cisplatin-resistant human epidermoid carcinoma tumor cells. J. Natl. Cancer Inst. 1994, 86, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Twentyman, P.R.; Wright, K.A.; Mistry, P.; Kelland, L.R.; Murrer, B.A. Sensitivity to novel platinum compounds of panels of human lung cancer cell lines with acquired and inherent resistance to cisplatin. Cancer Res. 1992, 52, 5674–5680. [Google Scholar]
- Granzow, C.; Kopun, M.; Krober, T. Riboflavin-mediated photosensitization of Vinca alkaloids distorts drug sensitivity assays. Cancer Res. 1995, 55, 4837–4843. [Google Scholar]
- Heuser, M.; Kopun, M.; Rittgen, W.; Granzow, C. Cytotoxicity determination without photochemical artifacts. Cancer Lett. 2005, 223, 57–66. [Google Scholar] [CrossRef]
- Mirski, S.E.; Gerlach, J.H.; Cole, S.P. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin. Cancer Res. 1987, 47, 2594–2598. [Google Scholar]
- Twentyman, P.R.; Fox, N.E.; Wright, K.A.; Bleehen, N.M. Derivation and preliminary characterisation of adriamycin resistant lines of human lung cancer cells. Br. J. Cancer 1986, 53, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Stordal, B.; Pavlakis, N.; Davey, R. Oxaliplatin for the treatment of cisplatin-resistant cancer: A systematic review. Cancer Treat. Rev. 2007, 33, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Rixe, O.; Ortuzar, W.; Alvarez, M.; Parker, R.; Reed, E.; Paull, K.; Fojo, T. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: Spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute’s Anticancer Drug Screen panel. Biochem. Pharmacol. 1996, 52, 1855–1865. [Google Scholar] [CrossRef]
- Fukuda, M.; Ohe, Y.; Kanzawa, F.; Oka, M.; Hara, K.; Saijo, N. Evaluation of novel platinum complexes, inhibitors of topoisomerase I and II in non-small cell lung cancer (NSCLC) sublines resistant to cisplatin. Anticancer Res. 1995, 15, 393–398. [Google Scholar]
- Hector, S.; Bolanowska-Higdon, W.; Zdanowicz, J.; Hitt, S.; Pendyala, L. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother. Pharmacol. 2001, 48, 398–406. [Google Scholar] [CrossRef]
- Zhang, D.; Ochi, N.; Takigawa, N.; Tanimoto, Y.; Chen, Y.; Ichihara, E.; Hotta, K.; Tabata, M.; Tanimoto, M.; Kiura, K. Establishment of pemetrexed-resistant non-small cell lung cancer cell lines. Cancer Lett. 2011, 309, 228–235. [Google Scholar] [CrossRef]
- Buckley, A.M.; Bibby, B.A.; Dunne, M.R.; Kennedy, S.A.; Davern, M.B.; Kennedy, B.N.; Maher, S.G.; O’Sullivan, J. Characterisation of an Isogenic Model of Cisplatin Resistance in Oesophageal Adenocarcinoma Cells. Pharmaceuticals 2019, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Okuda, T.; Holzer, A.; Howell, S.B. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Mol. Pharmacol. 2002, 62, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3, 1543–1549. [Google Scholar]
- Ivy, K.D.; Kaplan, J.H. A re-evaluation of the role of hCTR1, the human high-affinity copper transporter, in platinum-drug entry into human cells. Mol. Pharmacol. 2013, 83, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, B.H.; Thorsteinsdottir, U.A.; Lambert, I.H. Acquired cisplatin resistance in human ovarian A2780 cancer cells correlates with shift in taurine homeostasis and ability to volume regulate. Am. J. Physiol. Cell Physiol. 2014, 307, C1071–C1080. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, B.H.; Nielsen, D.; Thorsteinsdottir, U.A.; Hoffmann, E.K.; Lambert, I.H. Downregulation of LRRC8A protects human ovarian and alveolar carcinoma cells against Cisplatin-induced expression of p53, MDM2, p21Waf1/Cip1, and Caspase-9/-3 activation. Am. J. Physiol. Cell Physiol. 2016, 310, C857–C873. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, B.H.; Dam, C.S.; Sturup, S.; Lambert, I.H. Dual role of LRRC8A-containing transporters on cisplatin resistance in human ovarian cancer cells. J. Inorg. Biochem. 2016, 160, 287–295. [Google Scholar] [CrossRef]
- Lee, C.C.; Freinkman, E.; Sabatini, D.M.; Ploegh, H.L. The protein synthesis inhibitor blasticidin s enters mammalian cells via leucine-rich repeat-containing protein 8D. J. Biol. Chem. 2014, 289, 17124–17131. [Google Scholar] [CrossRef] [Green Version]
- Granzow, C.; Drings, P.; Kopun, M. Identifizierung von Chemosensitivität bei menschlichen Tumorzellen durch flavinschützende Tests in vitro. In Thoraxtumoren: Diagnostik-Staging-Gegenwärtiges Therapiekonzept; Drings, P., Vogt–Moykopf, I., Eds.; Springer: Berlin, Germany, 1998; pp. 328–332. [Google Scholar]
- Dunnett, C.W. A Multiple Comparison Procedure for Comparing Several Treatments with a Control. J. Am. Stat. Assoc. 1955, 50, 1096–1121. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting Subline | A24 wt | A24 wt | A24cisPt0.5 | A24cisPt1.0 | A24cisPt2.0 | A24cisPt4.0 |
|---|---|---|---|---|---|---|
| cisPt concentration exposed to [µM] | n. a. | 0.5 | 1.0 | 2.0 | 4.0 | 8.0 |
| Branching-off after (months) | n. a. | 2 | 2 | 3 | 4 | 12 |
| Resulting subline | n. a. | A24cisPt0.5 | A24cisPt1.0 | A24cisPt2.0 | A24cisPt4.0 | A24cisPt8.0 |
| IC50 cisPt at branching-off time | 0.46 ± 0.05 | 2.13 ± 0.62 | 2.97 ± 0.81 | 6.03 ± 1.39 | 9.09 ± 0.56 | 18.49 ± 1.96 |
| De-induction period (months) | n. a. | 3 | 3 | 7 | 7 | >7 |
| De-induced subline | n. a. | (D-)A24cisPt0.5 | (D-)A24cisPt1.0 | (D-)A24cisPt2.0 | (D-)A24cisPt4.0 | (D-)A24cisPt8.0 |
| IC50 cisPt [µM] | n. a. | 0.89 ± 0.22 | 2.53 ± 0.84 | 0.99 ± 0.07 | 4.02 ± 0.23 | 12.74 ± 0.78 |
| Population doubling time (h) | 20.5 | n. d. | n. d. | 25 | 31 | 42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruprecht, N.; Hofmann, L.; Hungerbühler, M.N.; Kempf, C.; Heverhagen, J.T.; von Tengg-Kobligk, H. Generation of Stable cisPt Resistant Lung Adenocarcinoma Cells. Pharmaceuticals 2020, 13, 109. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13060109
Ruprecht N, Hofmann L, Hungerbühler MN, Kempf C, Heverhagen JT, von Tengg-Kobligk H. Generation of Stable cisPt Resistant Lung Adenocarcinoma Cells. Pharmaceuticals. 2020; 13(6):109. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13060109
Chicago/Turabian StyleRuprecht, Nico, Lukas Hofmann, Martin Nils Hungerbühler, Christoph Kempf, Johannes Thomas Heverhagen, and Hendrik von Tengg-Kobligk. 2020. "Generation of Stable cisPt Resistant Lung Adenocarcinoma Cells" Pharmaceuticals 13, no. 6: 109. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13060109