
Population Pharmacokinetic Method to Predict Within-Subject Variability Using Single-Period Clinical Data

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
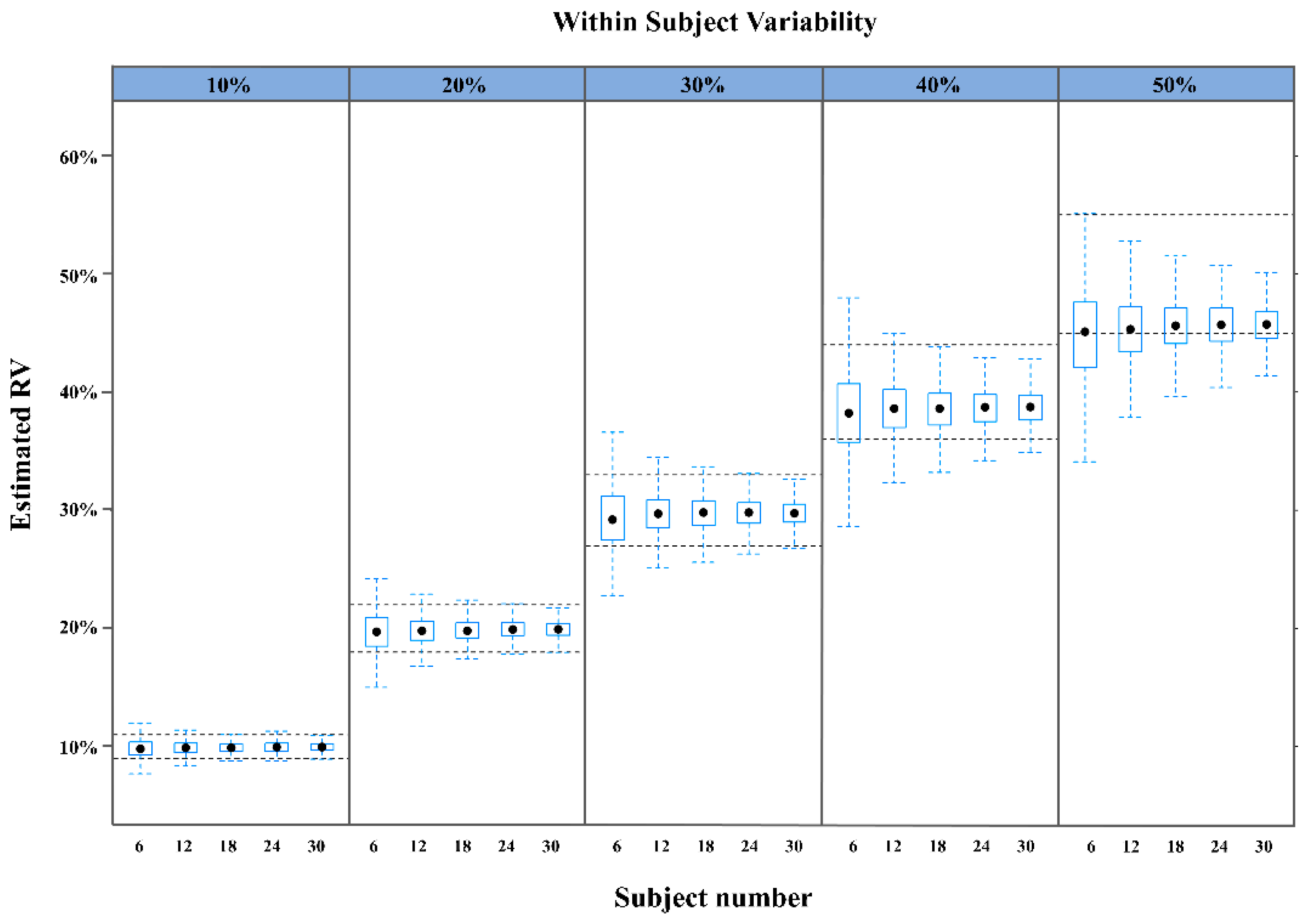
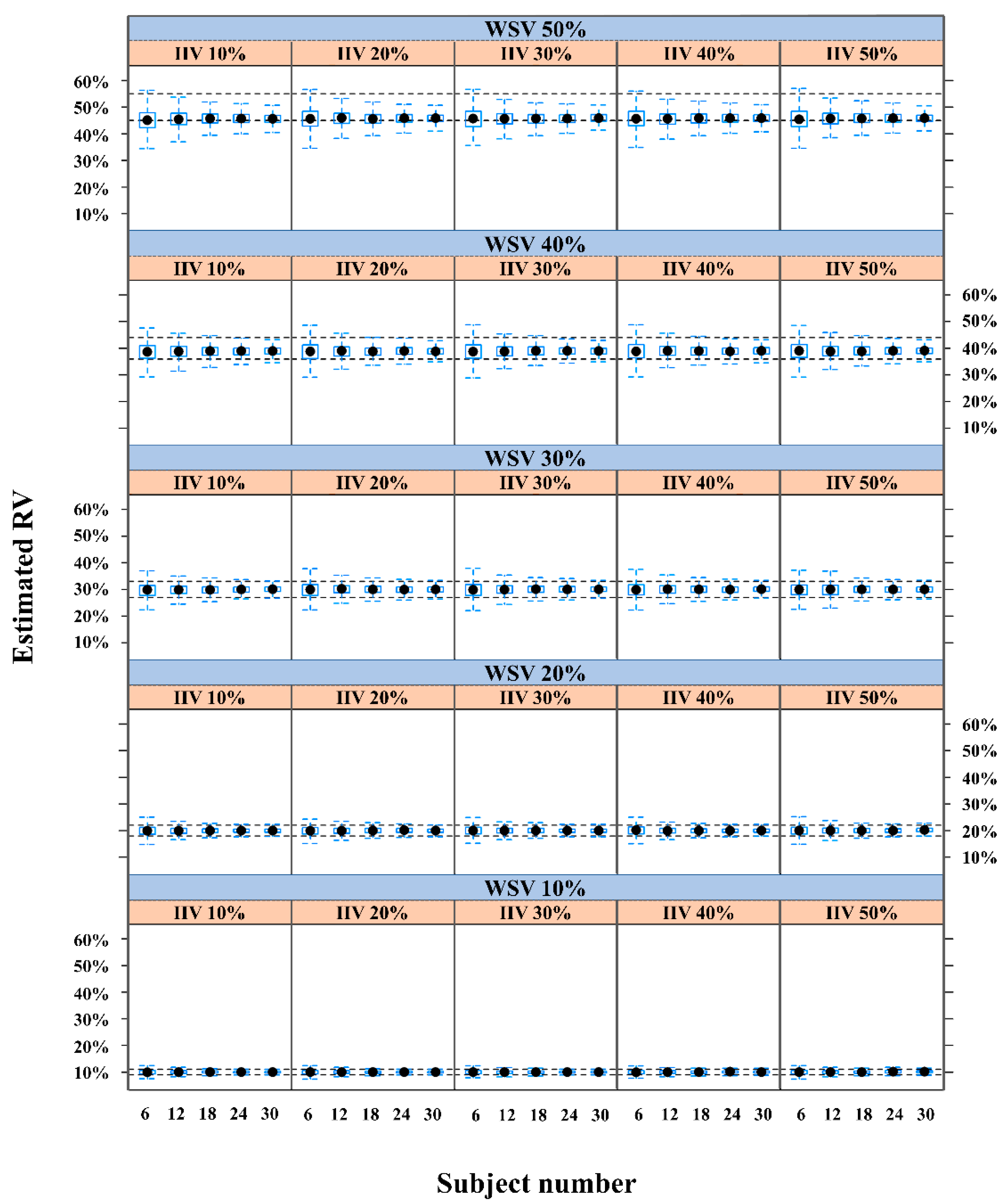
2.1. Performance of a Population-Based Method Using a Virtual Single-Period Clinical PK Trial
2.2. Real Case Application
3. Discussion
4. Materials and Methods
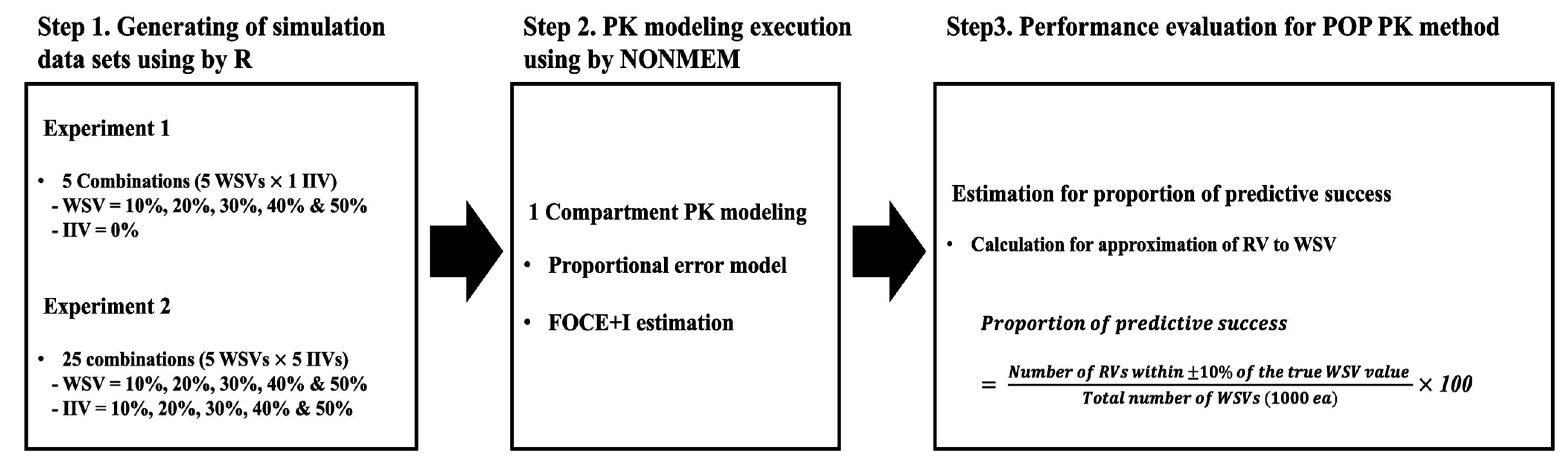
4.1. Overall Scheme to Evaluate Performance of the Population-Based Method
4.2. Generating a Simulation Dataset and Performing Population PK Modeling for Scenarios without Changes in IIV (First Experiment)
4.3. Generating Simulation Datasets and Performing Population PK Modeling for Scenarios with Differing WSVs and IIVs (Second Experiment)
4.4. Evaluation of Predictive Power
4.5. Application to Clinical Example (Real Case)
4.6. Softwares
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, K.K.; Attri, J.P.; Singh, A.; Kaur, H.; Kaur, G. Basic concepts for sample size calculation: Critical step for any clinical trials! Saudi J. Anaesth. 2016, 10, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ji, X. Sample Size Estimation in Clinical Research: From Randomized Controlled Trials to Observational Studies. Chest 2020, 158, S12–S20. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, B.; du Prel, J.B.; Wachtlin, D.; Kwiecien, R.; Blettner, M. Sample size calculation in clinical trials: Part 13 of a series on evaluation of scientific publications. Dtsch. Arztebl. Int. 2010, 107, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Sakpal, T.V. Sample size estimation in clinical trial. Perspect. Clin. Res. 2010, 1, 67–69. [Google Scholar] [PubMed]
- Center for Drug Evaluation and Research (CDER). Guidance for Industry: Population Pharmacokinetics; Food and Drug Administration (Draft): Silver Spring, MD, USA, 2019. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/population-pharmacokinetics (accessed on 11 January 2021).
- Mould, D.R.; Upton, R.N. Basic Concepts in Population Modeling, Simulation, and Model-Bsed Drug Development-Part 2: Introduction to Pharmacokinetic Modeling Method. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e38. [Google Scholar] [CrossRef] [PubMed]
- Manolache, M.; Cadar, E.; Antonescu, D.; Mircioiu, C.; Prasacu, I.; Sandulovici, R. Bioethics approach of biostatistics in clinical Trials. Avoid the use of excessive or inadequate numbers of research subjects. J. Sci. Arts 2018, 18, 239–246. [Google Scholar]
- Pradhan, S.; Song, B.; Lee, J.; Chae, J.; Kim, K.I.; Back, H.; Han, N.; Kwon, K.; Yun, H. Performance comparison of first-order conditional estimation with interaction and Bayesian estimation methods for estimating the population parameters and its distribution from data sets with a low number of subjects. BMC Med. Res. Methodol. 2017, 17, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, J.S.; Fiedler-Kelly, J. Introduction to Population Pharmacokinetic/Pharmacodynamic Analysis with Nonlinear Mixed Effects Model; Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 9–27. [Google Scholar]
- Fisher, D.; Shafer, S. Fisher/Shafer NONMEM Workshop Pharmacokinetics and Pharmacodynamic Analysis with Nonmem, Basic Concepts; Het Pand: Ghent, Belgium, 2007. [Google Scholar]
- Lee, H.J.; Ha, E.S.; Lee, Y.; Ha, D.C.; Choi, Y.W.; Baek, I.H.; Kim, M.S. Pharmacokinetic and bioequivalence study of sugar-coated and film-coated eprisone tablets in healthy subjects: A randomized, open-label, three-way, reference-replicated crossover study. Int. J. Clin. Pharmacol. Ther. 2019, 57, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Karalis, V.; Symilides, M.; Macheras, P. Bioequivalence of Highly Variable Drugs: A Comparison of the Newly Proposed Regulatory Approaches by FDA and EMA. Pharm. Res. 2012, 29, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence; European Medicines Agency: London, UK, 2010; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (accessed on 11 January 2021).
- Center for Drug Evaluation and Research (CDER). Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA; Food and Drug Administration: Silver Spring, MD, USA, 2013. Available online: https://www.fda.gov/files/drugs/published/Bioequivalence-Studies-With-Pharmacokinetic-Endpoints-for-Drugs-Submitted-Under-an-Abbreviated-New-Drug-Application.pdf (accessed on 11 January 2021).
- Haidar, S.H.; Davit, B.; Chen, M.L.; Conner, D.; Lee, L.; Li, Q.H.; Lionberger, R.; Makhlouf, F.; Patel, D.; Schuirmann, D.J.; et al. Bioequivalence Approaches for Highly Variable Drugs and Drug Products. Pharm. Res. 2008, 25, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Davit, B.M.; Conner, D.P.; Fabian-Fritsch, B.; Haidar, S.H.; Jiang, X.; Patel, D.T.; Seo, P.R.H.; Suh, K.; Thompson, C.L.; Yu, L.X. Highly Variable Drugs: Observations from Bioequivalence Data Submitted to the FDA for New Generic Drug Applications. AAPS J. 2008, 10, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkara, G.; Reynolds, C.V.; Pommier, F.; Humbert, H.; Yeh, C.; Prasad, P. Evaluation of a pharmacokinetic interaction between valsartan and simvastatin in healthy subjects. Curr. Med. Res. Opin. 2007, 23, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Tsang, Y.C.; Pop, R.; Gordon, P.; Hems, J.; Spino, M. High variability in drug pharmacokinetics complicates determination of bioequivalence: Experience with verapamil. Pharm. Res. 1996, 13, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Buice, R.G.; Subramanian, V.S.; Duchin, K.L. Bioequivalence of a Highly Variable Drug: An Experience with Nadolol. Pharm. Res. 1996, 13, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Blume, H.; Zhong, D.; Elze, M. Advantages of a steady-state crossover design in assessment of bioequivalence of highly variable drugs: Propafenone. Eur. J. Pharm. Sci. 1994, 2, 385–393. [Google Scholar] [CrossRef]
- Huang, Y.C.; Colaizzi, J.L.; Bierman, R.H.; Woestenborghs, R.; Heykants, J. Pharmacokinetics and dose proportionality of ketoconazole in normal volunteers. Antimicrob. Agents Chemother. 1986, 30, 206–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alakhali, K.; Hassan, Y.; Mohamed, N.; Mordi, M.N. Pharmacokinetic of simvastatin study in Malaysian subjects. IOSR J. Pharm. 2013, 3, 46–51, ISSN: 2250–3013. [Google Scholar] [CrossRef]
- Prescribing Information of Voltaren. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/019201s038lbl.pdf (accessed on 23 January 2021).
- Sherwin, C.M.; Saldaña, S.N.; Bies, R.R.; Aman, M.G.; Vinks, A.A. Population pharmacokinetic modeling of risperidone and 9-hydroxyrisperidone to estimate CYP2D6 subpopulations in children and adolescents. Ther. Drug Monit. 2012, 34, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Yin, Q.; Deckert, F.; Jiang, J.; Liu, D.; Kjems, L.; Dole, W.P.; He, Y.L. Pharmacokinetics and pharmacodynamics of vildagliptin in healthy Chinese volunteers. J. Clin. Pharmacol. 2009, 49, 39–49. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| WSV (%) | Proportion of Predictive Success (%) * for Each Subject Number | ||||
|---|---|---|---|---|---|
| n = 6 | n = 12 | n = 18 | n = 24 | n = 30 | |
| 10 | 75 | 91 | 95 | 99 | 99 |
| 20 | 75 | 91 | 96 | 98 | 99 |
| 30 | 71 | 90 | 96 | 98 | 99 |
| 40 | 66 | 84 | 90 | 94 | 96 |
| 50 | 51 | 53 | 62 | 63 | 66 |
| Setting Condition | Proportion of Predictive Success (%)* for Each Subject Number | |||||
|---|---|---|---|---|---|---|
| WSV (%) | IIV (%) | n = 6 | n = 12 | n = 18 | n = 24 | n = 30 |
| 10 | 10 | 72 | 85 | 94 | 97 | 99 |
| 20 | 68 | 87 | 94 | 98 | 99 | |
| 30 | 73 | 86 | 94 | 96 | 97 | |
| 40 | 69 | 87 | 90 | 92 | 93 | |
| 50 | 70 | 82 | 88 | 88 | 85 | |
| 20 | 10 | 70 | 86 | 93 | 96 | 98 |
| 20 | 74 | 87 | 94 | 97 | 99 | |
| 30 | 71 | 87 | 94 | 96 | 98 | |
| 40 | 70 | 88 | 95 | 95 | 99 | |
| 50 | 72 | 86 | 87 | 97 | 98 | |
| 30 | 10 | 73 | 88 | 93 | 98 | 99 |
| 20 | 70 | 86 | 92 | 96 | 98 | |
| 30 | 68 | 88 | 92 | 96 | 98 | |
| 40 | 69 | 84 | 94 | 96 | 98 | |
| 50 | 71 | 72 | 91 | 96 | 97 | |
| 40 | 10 | 71 | 82 | 89 | 94 | 98 |
| 20 | 66 | 83 | 90 | 95 | 96 | |
| 30 | 70 | 84 | 92 | 94 | 96 | |
| 40 | 70 | 87 | 91 | 94 | 97 | |
| 50 | 71 | 85 | 90 | 94 | 97 | |
| 50 | 10 | 50 | 56 | 62 | 62 | 63 |
| 20 | 54 | 62 | 59 | 66 | 65 | |
| 30 | 55 | 58 | 61 | 64 | 68 | |
| 40 | 56 | 59 | 65 | 65 | 67 | |
| 50 | 54 | 60 | 62 | 66 | 68 | |
| Single Period Dataset * | Measured Dataset [11] | |
|---|---|---|
| No. of Subjects | RV (%) | CVw for Cmax (%) |
| 6 | 45 | 50.21 |
| 12 | 48 | |
| 18 | 45 | |
| 24 | 44 | |
| 30 | 47 | |
| No. of Experiment | Scenario | Contents | Setting Value | ||||
|---|---|---|---|---|---|---|---|
| 1st | 1 | WSV (%) | 10 | 20 | 30 | 40 | 50 |
| IIV (%) | 0 | ||||||
| No. of subjects | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | ||
| 2nd | 1 | WSV (%) | 10 | ||||
| IIV (%) | 10 | 20 | 30 | 40 | 50 | ||
| No. of subjects | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | ||
| 2 | WSV (%) | 20 | |||||
| IIV (%) | 10 | 20 | 30 | 40 | 50 | ||
| No. of subjects | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | ||
| 3 | WSV (%) | 30 | |||||
| IIV (%) | 10 | 20 | 30 | 40 | 50 | ||
| No. of subjects | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | ||
| 4 | WSV (%) | 40 | |||||
| IIV (%) | 10 | 20 | 30 | 40 | 50 | ||
| No. of subjects | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | ||
| 5 | WSV (%) | 50 | |||||
| IIV (%) | 10 | 20 | 30 | 40 | 50 | ||
| No. of subjects | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | 6→12→18→24→30 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, W.-h.; Lee, J.-y.; Chae, J.-w.; Lee, K.-R.; Baek, I.-h.; Kim, M.-S.; Back, H.-m.; Jung, S.; Shaffer, C.; Savic, R.; et al. Population Pharmacokinetic Method to Predict Within-Subject Variability Using Single-Period Clinical Data. Pharmaceuticals 2021, 14, 114. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020114
Kang W-h, Lee J-y, Chae J-w, Lee K-R, Baek I-h, Kim M-S, Back H-m, Jung S, Shaffer C, Savic R, et al. Population Pharmacokinetic Method to Predict Within-Subject Variability Using Single-Period Clinical Data. Pharmaceuticals. 2021; 14(2):114. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020114
Chicago/Turabian StyleKang, Won-ho, Jae-yeon Lee, Jung-woo Chae, Kyeong-Ryoon Lee, In-hwan Baek, Min-Soo Kim, Hyun-moon Back, Sangkeun Jung, Craig Shaffer, Rada Savic, and et al. 2021. "Population Pharmacokinetic Method to Predict Within-Subject Variability Using Single-Period Clinical Data" Pharmaceuticals 14, no. 2: 114. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020114