
Discovery and Optimization of Selective Inhibitors of Meprin α (Part I)

 , ,
, ,
Abstract
:1. Introduction
2. Results
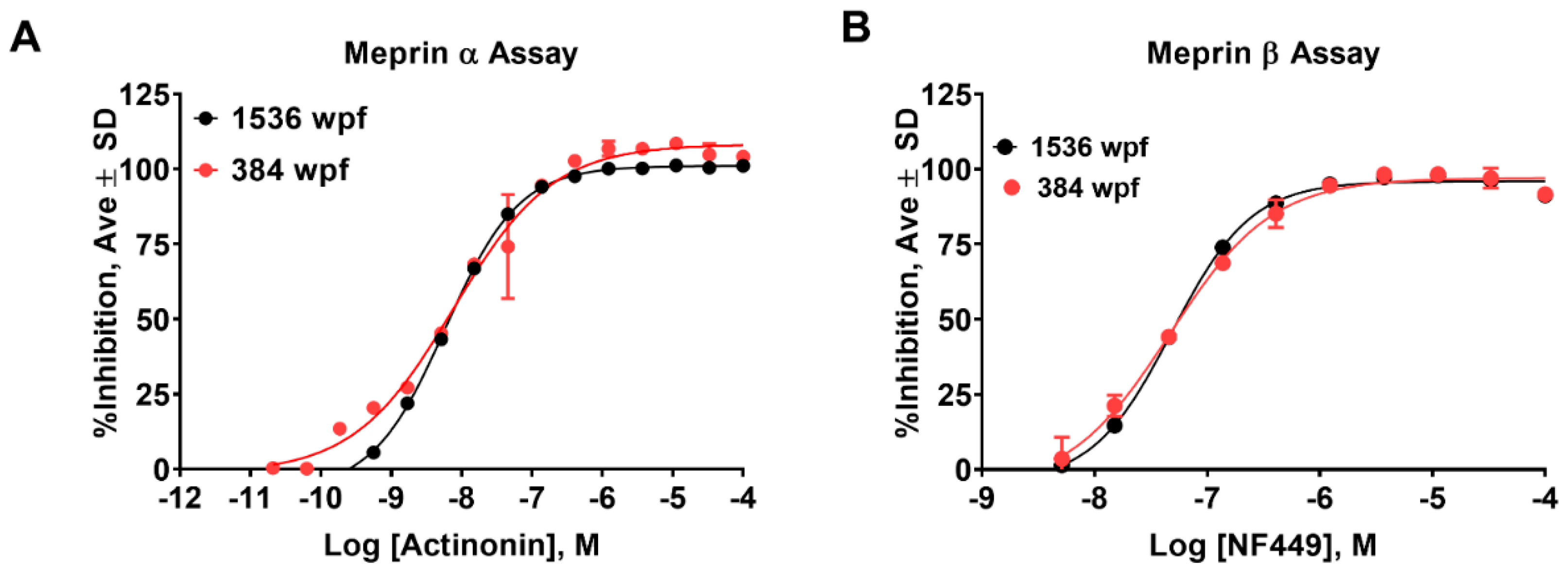
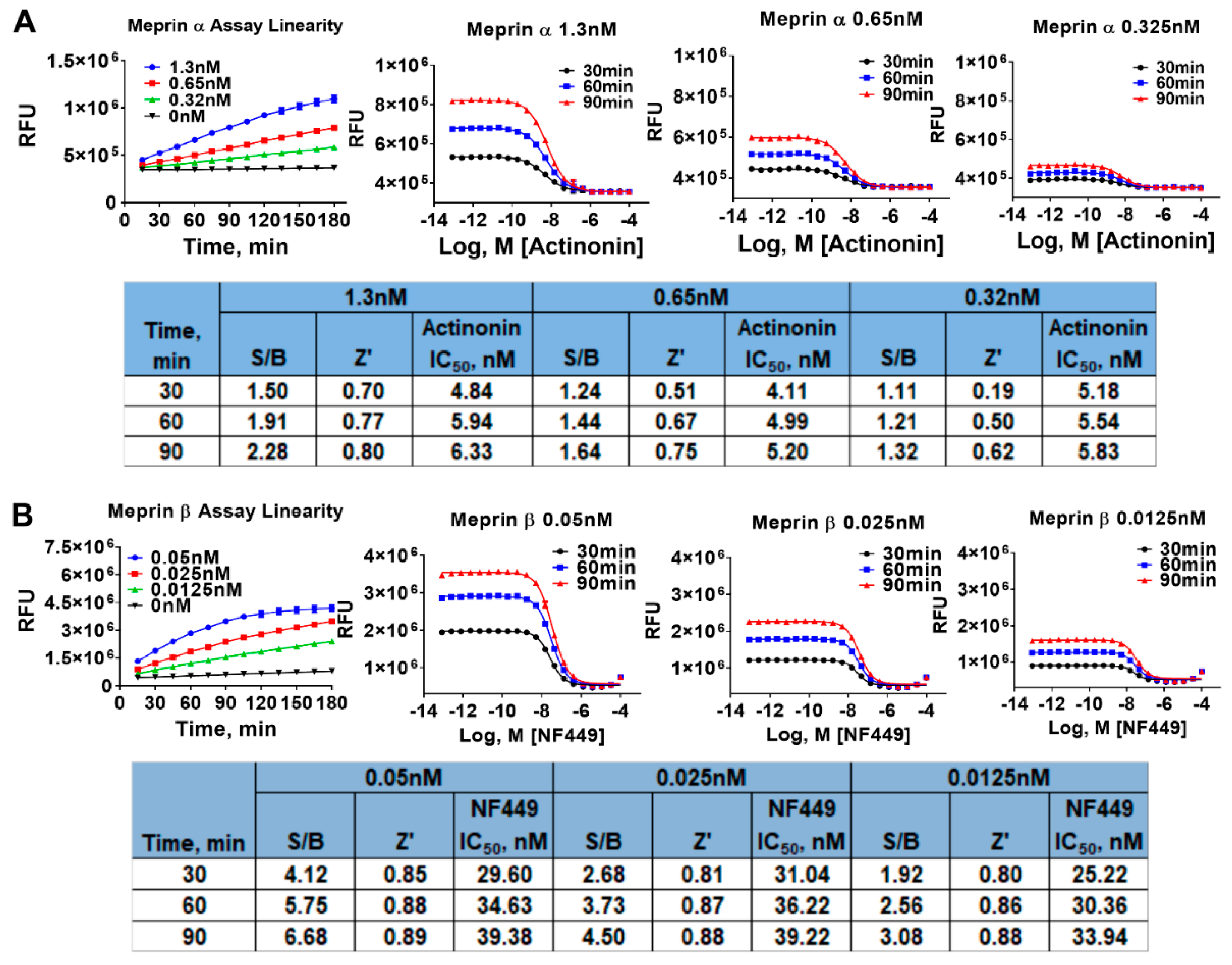
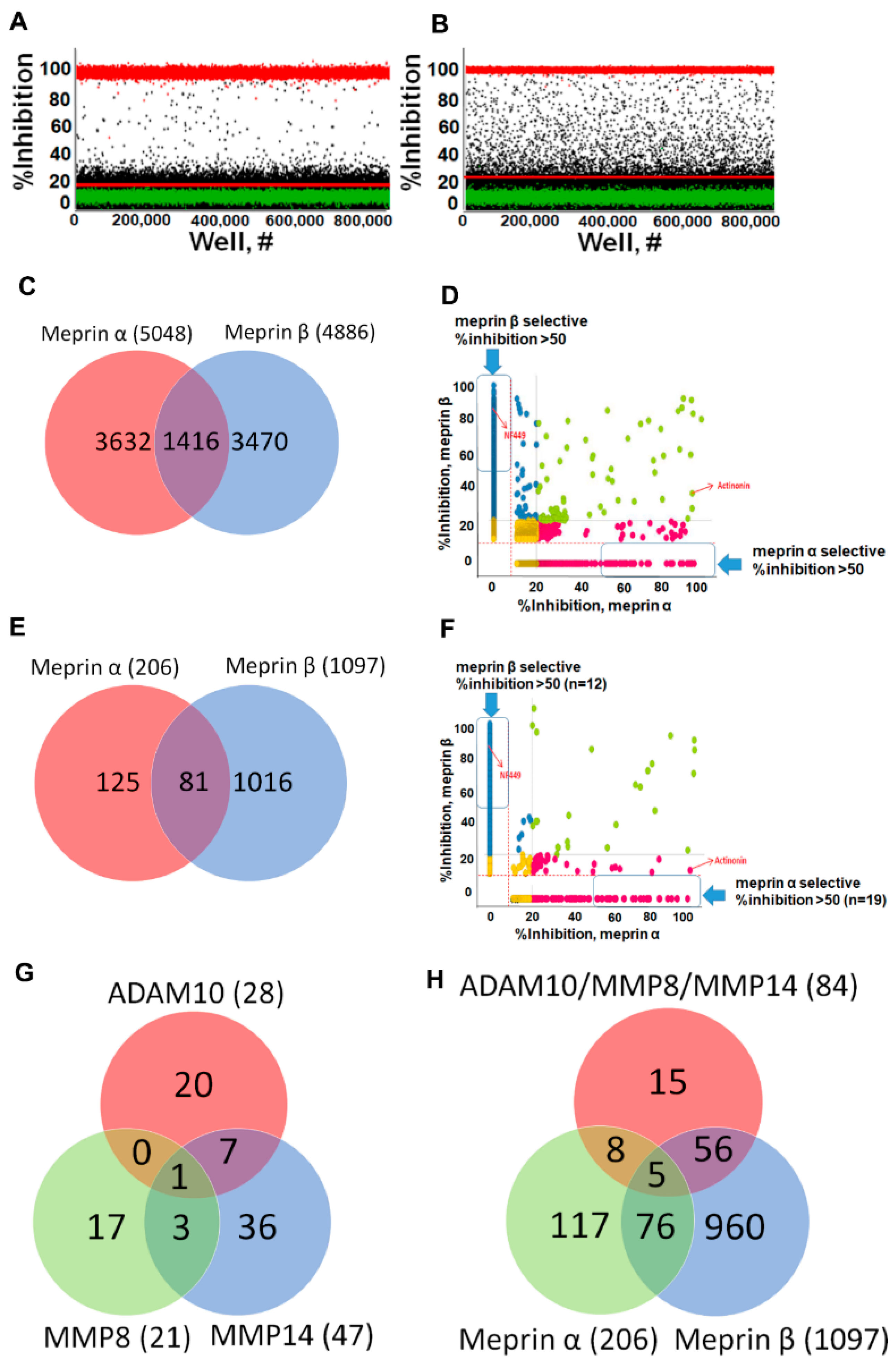
2.1. Assay Miniaturization and Optimization in 1536 Well Plate Format
2.2. Online Robotic Pilot Study
2.3. Primary HTS Campaign
2.4. Hit Confirmation and Prioritization
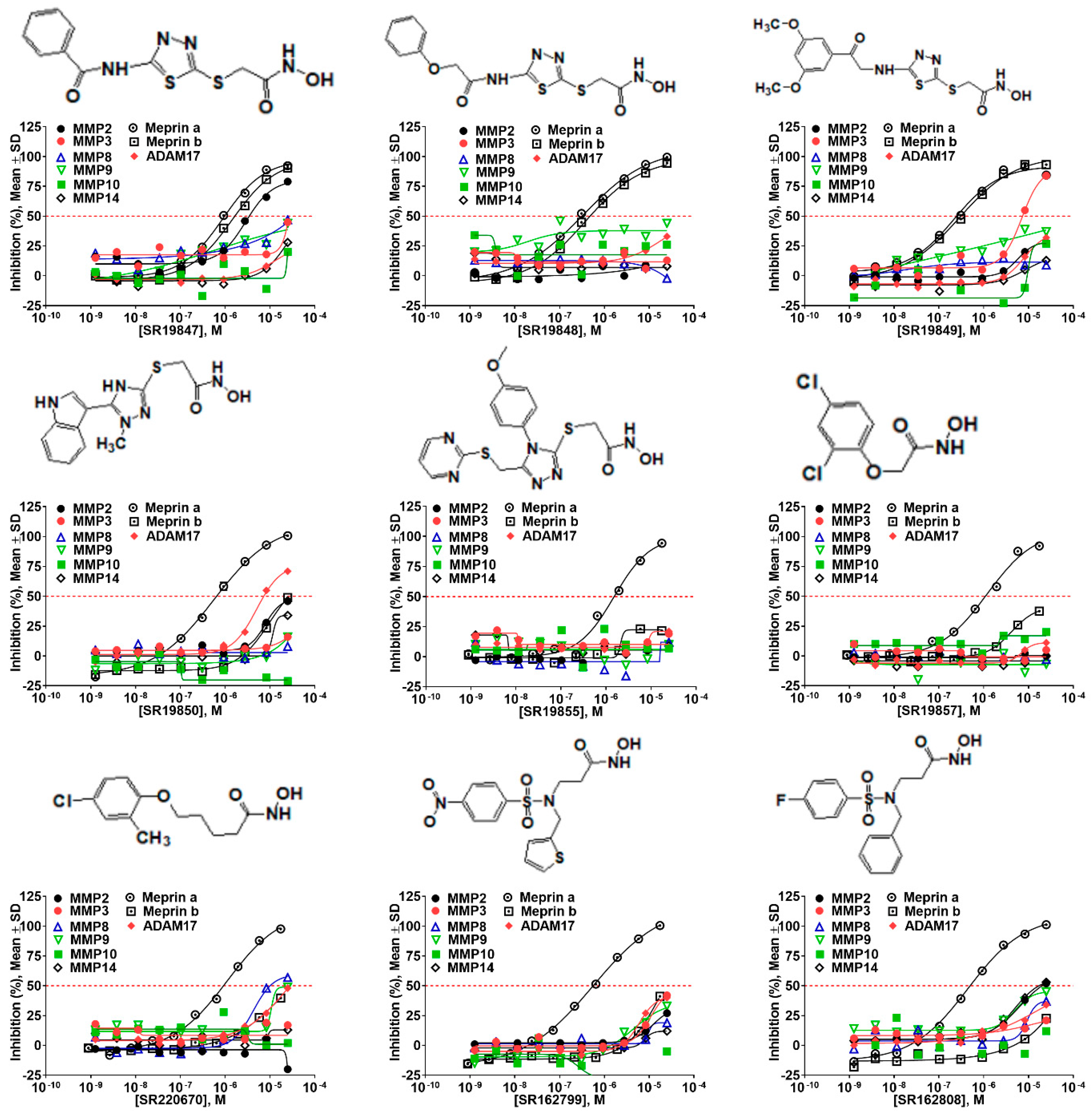
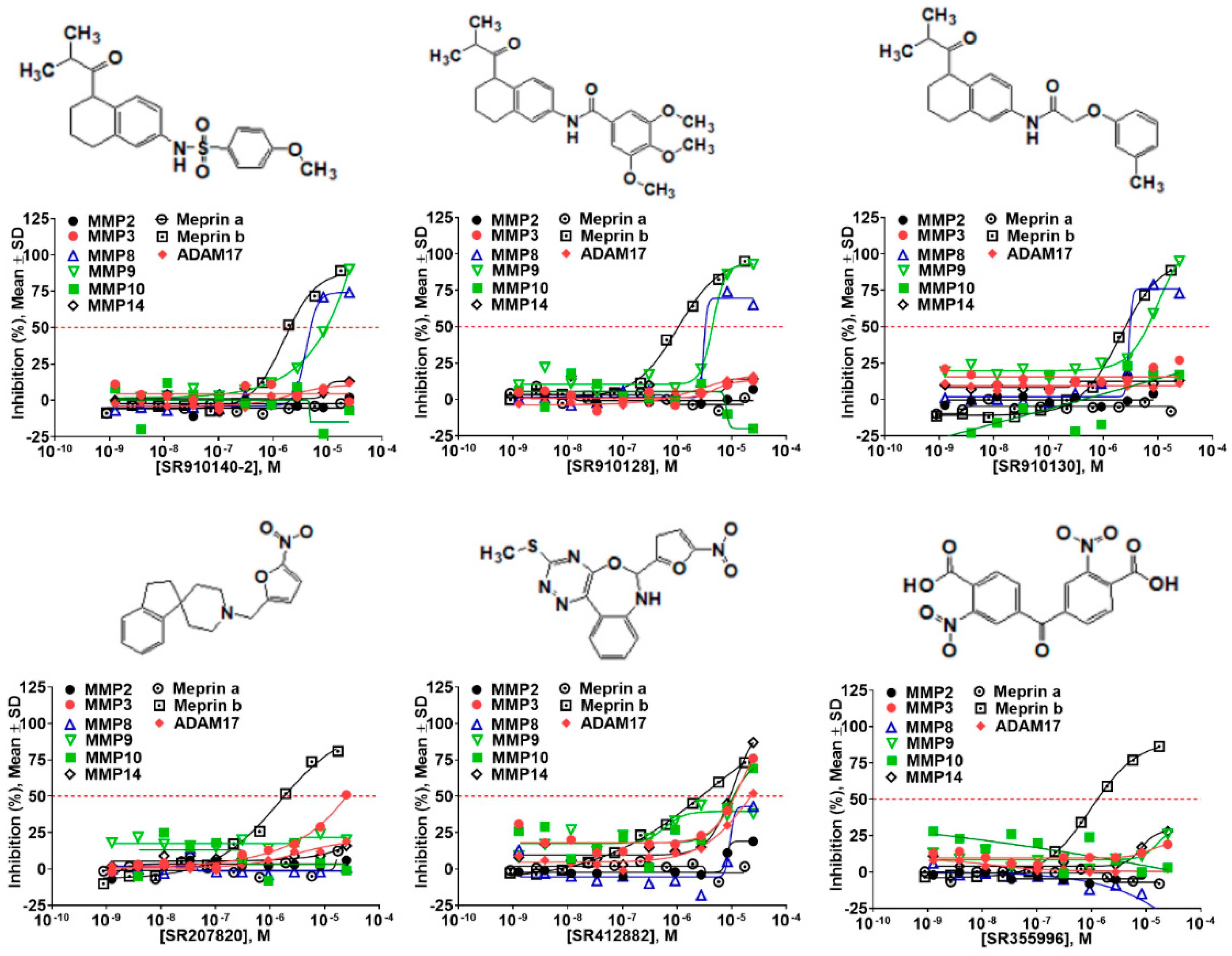
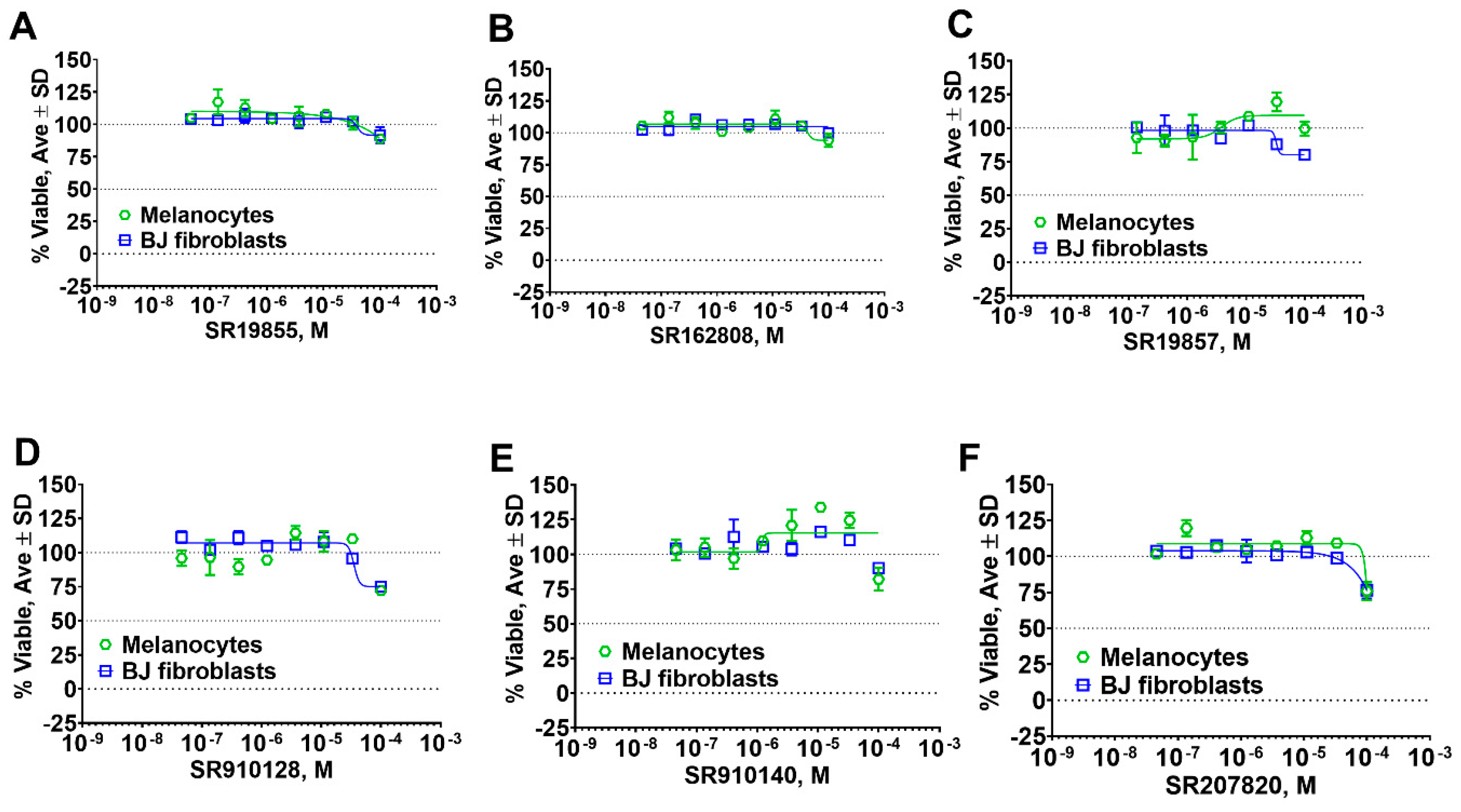
2.5. Hit Potency, Selectivity, and Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. HTS Substrate Synthesis
4.3. Meprins Expression Protocol
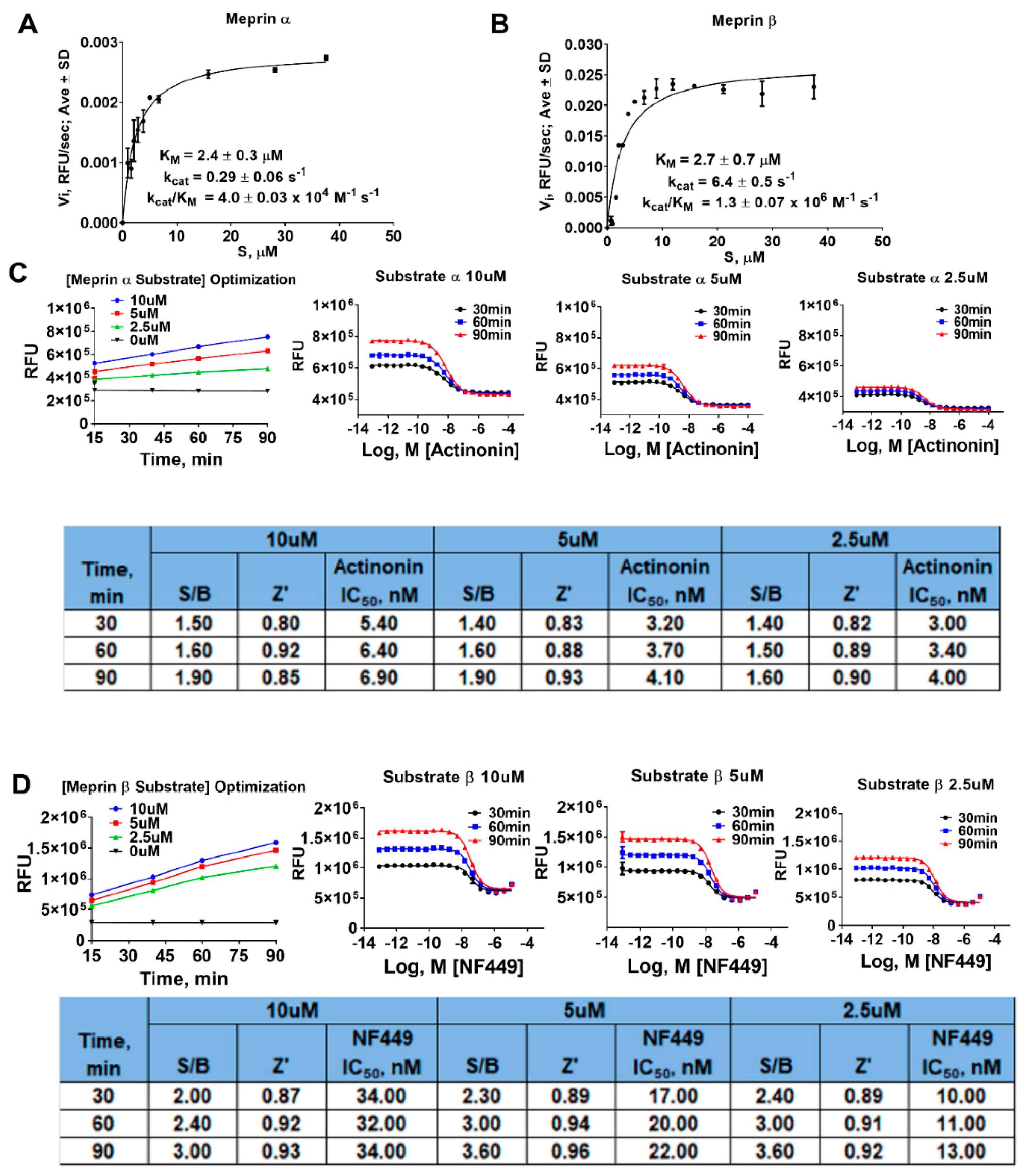
4.4. Meprin α and Meprin β Assays in 384 Well Plate
4.5. Determination of Kinetic Parameters of Meprin α and Meprin β Mediated Proteolysis of Their Respective Substrates
4.6. Meprin α and Meprin β Assays in 1536 Well Plate Format
4.7. uHTS Campaign
4.8. ADAM10 and ADAM17 Assays
4.9. MMP Assays
4.10. Cell Toxicity Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| uHTS | ultrahigh-throughput screening |
| MMP | matrix metalloprotease |
| ADAM | a disintegrin and metalloprotease |
References
- Peters, F.; Becker-Pauly, C. Role of meprin metalloproteases in metastasis and tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 347–356. [Google Scholar] [CrossRef]
- Prox, J.; Arnold, P.; Becker-Pauly, C. Meprin α and meprin β: Procollagen proteinases in health and disease. Matrix Biol. 2015, 44, 7–13. [Google Scholar] [CrossRef]
- Broder, C.; Arnold, P.; Vadon-Le Goff, S.; Konerding, M.A.; Bahr, K.; Muller, S.; Overall, C.M.; Bond, J.S.; Koudelka, T.; Tholey, A.; et al. Metalloproteases meprin alpha and meprin β are C- and N-procollagen proteinases important for collagen assembly and tensile strength. Proc. Natl. Acad. Sci. USA 2013, 110, 14219–14224. [Google Scholar] [CrossRef] [Green Version]
- Becker-Pauly, C.; Pietrzik, C.U. The Metalloprotease Meprin β Is an Alternative β-Secretase of APP. Front. Mol. Neurosci. 2016, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Scharfenberg, F.; Armbrust, F.; Marengo, L.; Pietrzik, C.; Becker-Pauly, C. Regulation of the alternative β-secretase meprin β by ADAM-mediated shedding. Cell. Mol. Life Sci. 2019, 76, 3193–3206. [Google Scholar] [CrossRef] [PubMed]
- Ohler, A.; Debela, M.; Wagner, S.; Magdolen, V.; Becker-Pauly, C. Analyzing the protease web in skin: Meprin metalloproteases are activated specifically by KLK4, 5 and 8 vice versa leading to processing of proKLK7 thereby triggering its activation. Biol. Chem. 2010, 391, 455–460. [Google Scholar] [CrossRef]
- Bhogal, R.K.; Stoica, C.M.; McGaha, T.L.; Bona, C.A. Molecular aspects of regulation of collagen gene expression in fibrosis. J. Clin. Immunol. 2005, 25, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Trojanowska, M.; LeRoy, E.C.; Eckes, B.; Krieg, T. Pathogenesis of fibrosis: Type 1 collagen and the skin. J. Mol. Med. 1998, 76, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Biasin, V.; Wygrecka, M.; Marsh, L.M.; Becker-Pauly, C.; Brcic, L.; Ghanim, B.; Klepetko, W.; Olschewski, A.; Kwapiszewska, G. Meprin β contributes to collagen deposition in lung fibrosis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, P.; Otte, A.; Becker-Pauly, C. Meprin metalloproteases: Molecular regulation and function in inflammation and fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2096–2104. [Google Scholar] [CrossRef]
- Guidi, A.; Mansour, N.R.; Paveley, R.A.; Carruthers, I.M.; Besnard, J.; Hopkins, A.L.; Gilbert, I.H.; Bickle, Q.D. Application of RNAi to Genomic Drug Target. Validation in Schistosomes. PLoS Negl. Trop. Dis. 2015, 9, e0003801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farand, J.; Kropf, J.E.; Blomgren, P.; Xu, J.; Schmitt, A.C.; Newby, Z.E.; Wang, T.; Murakami, E.; Barauskas, O.; Sudhamsu, J.; et al. Discovery of Potent and Selective MTH1 Inhibitors for Oncology: Enabling Rapid Target (In)Validation. ACS Med. Chem. Lett. 2019, 11, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.N.; Becker, C.; Lottaz, D.; Kohler, D.; Yiallouros, I.; Krell, H.W.; Sterchi, E.E.; Stocker, W. Human meprin α and β homo-oligomers: Cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem. J. 2004, 378, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
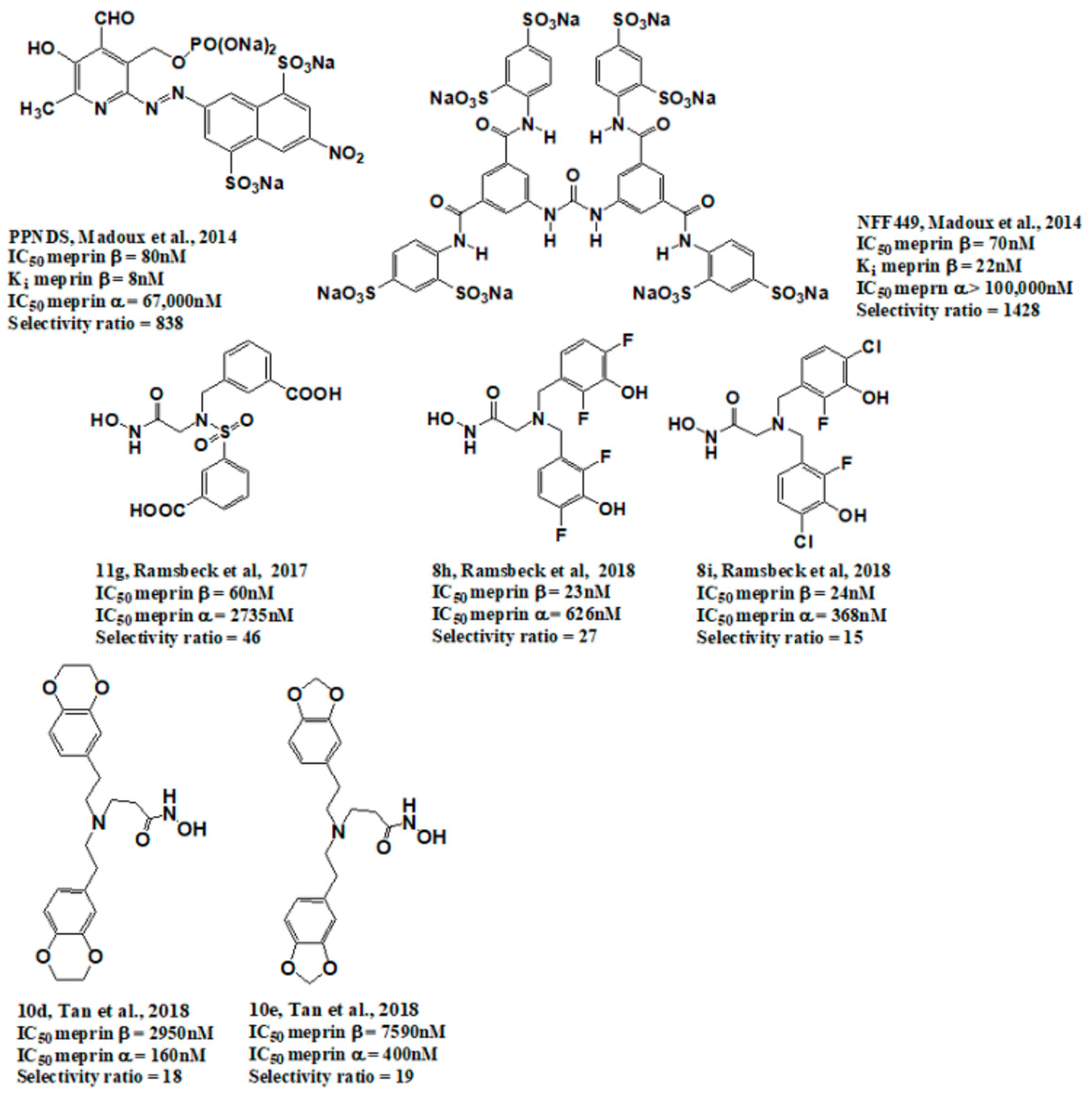
- Madoux, F.; Tredup, C.; Spicer, T.P.; Scampavia, L.; Chase, P.S.; Hodder, P.S.; Fields, G.B.; Becker-Pauly, C.; Minond, D. Development of high throughput screening assays and pilot screen for inhibitors of metalloproteases meprin α and β. Biopolymers 2014, 102, 396–406. [Google Scholar] [CrossRef] [Green Version]
- Ramsbeck, D.; Hamann, A.; Schlenzig, D.; Schilling, S.; Buchholz, M. First insight into structure-activity relationships of selective meprin β inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2428–2431. [Google Scholar] [CrossRef] [PubMed]
- Ramsbeck, D.; Hamann, A.; Richter, G.; Schlenzig, D.; Geissler, S.; Nykiel, V.; Cynis, H.; Schilling, S.; Buchholz, M. Structure-Guided Design, Synthesis, and Characterization of Next-Generation Meprin β Inhibitors. J. Med. Chem. 2018, 61, 4578–4592. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Jager, C.; Schlenzig, D.; Schilling, S.; Buchholz, M.; Ramsbeck, D. Tertiary-Amine-Based Inhibitors of the Astacin Protease Meprin alpha. ChemMedChem 2018, 13, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High. Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R. Evaluation of Enzyme Inhibitors in Drug Discovery, 1st ed.; John Wiley and Sons: Hoboken, NJ, USA, 2005; pp. 113–117. [Google Scholar]
- Madoux, F.; Dreymuller, D.; Pettiloud, J.P.; Santos, R.; Becker-Pauly, C.; Ludwig, A.; Fields, G.B.; Bannister, T.; Spicer, T.P.; Cudic, M.; et al. Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 2016, 6, 1–17. [Google Scholar] [CrossRef]
- Baillargeon, P.; Fernandez-Vega, V.; Sridharan, B.P.; Brown, S.; Griffin, P.R.; Rosen, H.; Cravatt, B.; Scampavia, L.; Spicer, T.P. The Scripps Molecular Screening Center and Translational Research Institute. SLAS Discov. Adv. Life Sci. R D 2019, 24, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liang, Y.; Si, X. Hydroxamic acid hybrids as the potential anticancer agents: An Overview. Eur. J. Med. Chem. 2020, 205, 112679. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Gao, R.; Dang, B.; Chen, G. The Blood Component Iron Causes Neuronal Apoptosis Following Intracerebral Hemorrhage via the PERK Pathway. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Broder, C.; Becker-Pauly, C. The metalloproteases meprin α and meprin β: Unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem. J. 2013, 450, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, G.B.; Lauer-Fields, J.L.; Liu, R.-q.; Barany, G. Principles and Practice of Solid-Phase Peptide Synthesis. In Synthetic Peptides: A User’s Guide, 2nd ed.; Grant, G.A., Ed.; W.H. Freeman & Co.: New York, NY, USA, 2001; pp. 93–219. [Google Scholar]
- de Jong, G.I.; Buwalda, B.; Schuurman, T.; Luiten, P.G. Synaptic plasticity in the dentate gyrus of aged rats is altered after chronic nimodipine application. Brain Res. 1992, 596, 345–348. [Google Scholar] [CrossRef] [Green Version]
- Becker-Pauly, C.; Howel, M.; Walker, T.; Vlad, A.; Aufenvenne, K.; Oji, V.; Lottaz, D.; Sterchi, E.E.; Debela, M.; Magdolen, V.; et al. The alpha and beta subunits of the metalloprotease meprin are expressed in separate layers of human epidermis, revealing different functions in keratinocyte proliferation and differentiation. J. Investig. Dermatol. 2007, 127, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Kruse, M.N.; Slotty, K.A.; Kohler, D.; Harris, J.R.; Rosmann, S.; Sterchi, E.E.; Stocker, W. Differences in the activation mechanism between the alpha and beta subunits of human meprin. Biol. Chem. 2003, 384, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Chase, P.; Niswender, C.M.; Utley, T.J.; Sheffler, D.J.; Noetzel, M.J.; Lamsal, A.; Wood, M.R.; Conn, P.J.; Lindsley, C.W.; et al. Application of Parallel Multiparametric Cell-Based FLIPR Detection Assays for the Identification of Modulators of the Muscarinic Acetylcholine Receptor 4 (M4). J. Biomol. Screen. 2015, 20, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | S/B | Z’ | Actinonin IC50, nM | NFF449 IC50, nM |
|---|---|---|---|---|
| Meprin α 384 wpf | 2.3 | 0.6 | 11 | >100,000 |
| Meprin α 1536 wpf | 1.85 | 0.76 | 5.7 | >100,000 |
| Meprin β 384 wpf | 4.4 | 0.9 | 22,000 | 53 |
| Meprin β 1536 wpf | 6.9 | 0.91 | 9750 | 48 |
| Compound ID | Structure | Meprin α | Meprin β | MMP2 | MMP3 | MMP8 | MMP9 | MMP10 | MMP14 | ADAM17 |
|---|---|---|---|---|---|---|---|---|---|---|
| 19847 |  | 0.892 | 1.43 | 2.87 | >17 | >17 | >17 | >17 | >17 | >17 |
| 19848 |  | 0.335 | 0.385 | >17 | >17 | >17 | >17 | >17 | >17 | >17 |
| 19849 |  | 0.218 | 0.287 | >17 | >17 | >17 | >17 | >17 | >17 | 8.01 |
| 19850 |  | 0.564 | 17 | 17 | >17 | >17 | >17 | >17 | >17 | 5.03 |
| 19855 |  | 1.3 | >17 | >17 | >17 | >17 | >17 | >17 | >17 | >17 |
| 1596857 |  | 1.18 | >17 | >17 | >17 | >17 | >17 | >17 | >17 | >17 |
| 220670 |  | 1.12 | >17 | >17 | >17 | 4.20 | 17 | >17 | >17 | >17 |
| 162799 |  | 0.564 | >17 | >17 | >17 | >17 | >17 | >17 | >17 | >17 |
| 162808 |  | 0.446 | >17 | 17 | >17 | >17 | >17 | >17 | >17 | >17 |
| Compound ID | Structure | Meprin α | Meprin β | MMP-2 | MMP-3 | MMP-8 | MMP-9 | MMP-10 | MMP-14 | ADAM17 |
|---|---|---|---|---|---|---|---|---|---|---|
| SR207820 |  | >17 | 1.5 | >17 | 17 | >17 | >17 | >17 | >17 | >17 |
| SR412882 |  | >17 | 3.5 | >17 | 15 | >17 | >17 | 9.7 | 10.5 | 17 |
| SR910128 |  | >17 | 1.0 | >17 | >17 | 3.1 | 4.5 | >17 | >17 | >17 |
| SR910130 |  | >17 | 2.0 | >17 | >17 | 3.0 | 9.9 | >17 | >17 | >17 |
| SR910140 |  | >17 | 1.6 | >17 | >17 | 4.0 | 10 | >17 | >17 | >17 |
| SR355996 |  | >17 | 0.97 | >17 | >17 | >17 | 17 | >17 | >17 | >17 |
| ID | Meprin α | Meprin β | Selectivity Fold |
|---|---|---|---|
| SR162808 | 0.30 | >17 | 38 |
| 10d | 0.16 | 2.95 | 18 |
| 10e | 0.40 | 7.59 | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, S.; Diez, J.; Wang, C.; Becker-Pauly, C.; Fields, G.B.; Bannister, T.; Spicer, T.P.; Scampavia, L.D.; Minond, D. Discovery and Optimization of Selective Inhibitors of Meprin α (Part I). Pharmaceuticals 2021, 14, 203. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030203
Hou S, Diez J, Wang C, Becker-Pauly C, Fields GB, Bannister T, Spicer TP, Scampavia LD, Minond D. Discovery and Optimization of Selective Inhibitors of Meprin α (Part I). Pharmaceuticals. 2021; 14(3):203. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030203
Chicago/Turabian StyleHou, Shurong, Juan Diez, Chao Wang, Christoph Becker-Pauly, Gregg B. Fields, Thomas Bannister, Timothy P. Spicer, Louis D. Scampavia, and Dmitriy Minond. 2021. "Discovery and Optimization of Selective Inhibitors of Meprin α (Part I)" Pharmaceuticals 14, no. 3: 203. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030203