
Dexamethasone Induces the Expression and Function of Tryptophan-2-3-Dioxygenase in SK-MEL-28 Melanoma Cells



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
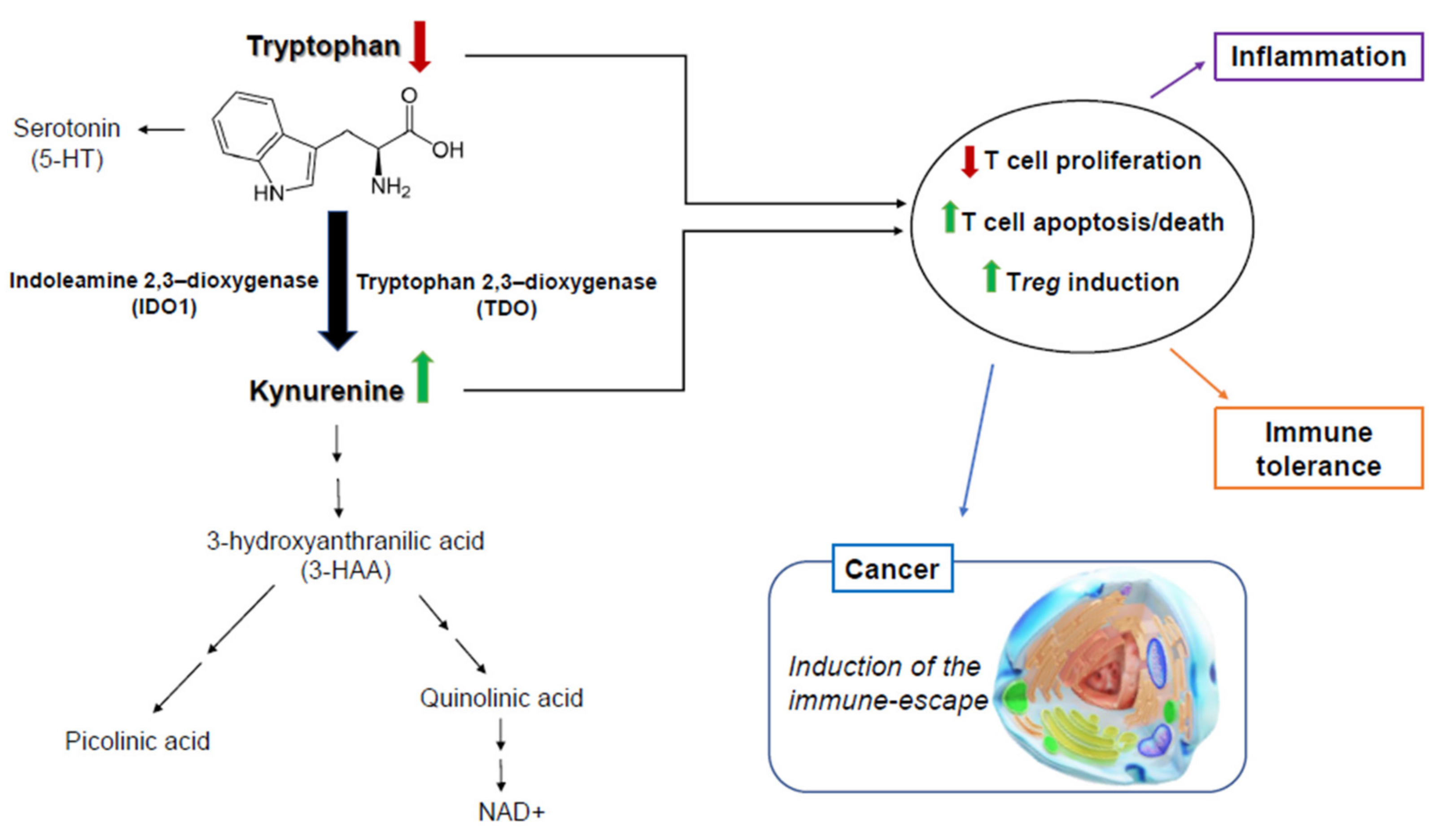
1. Introduction
2. Results
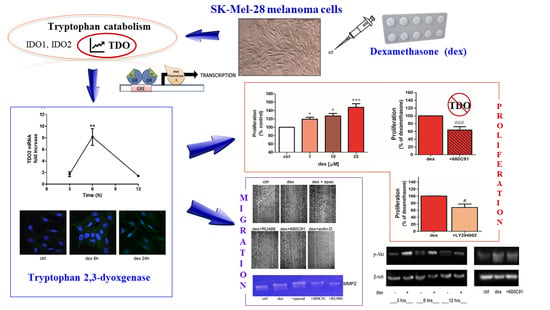
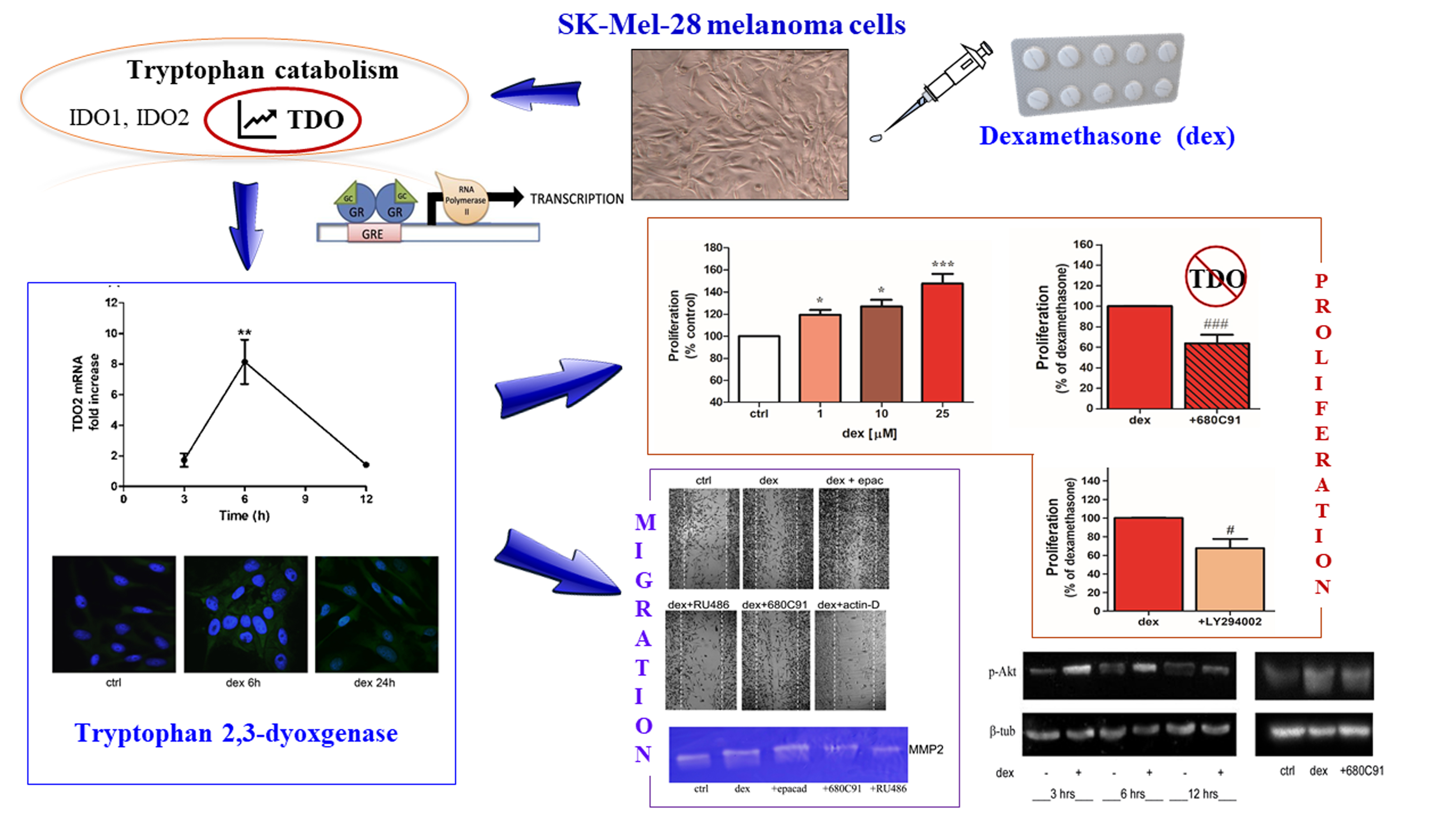
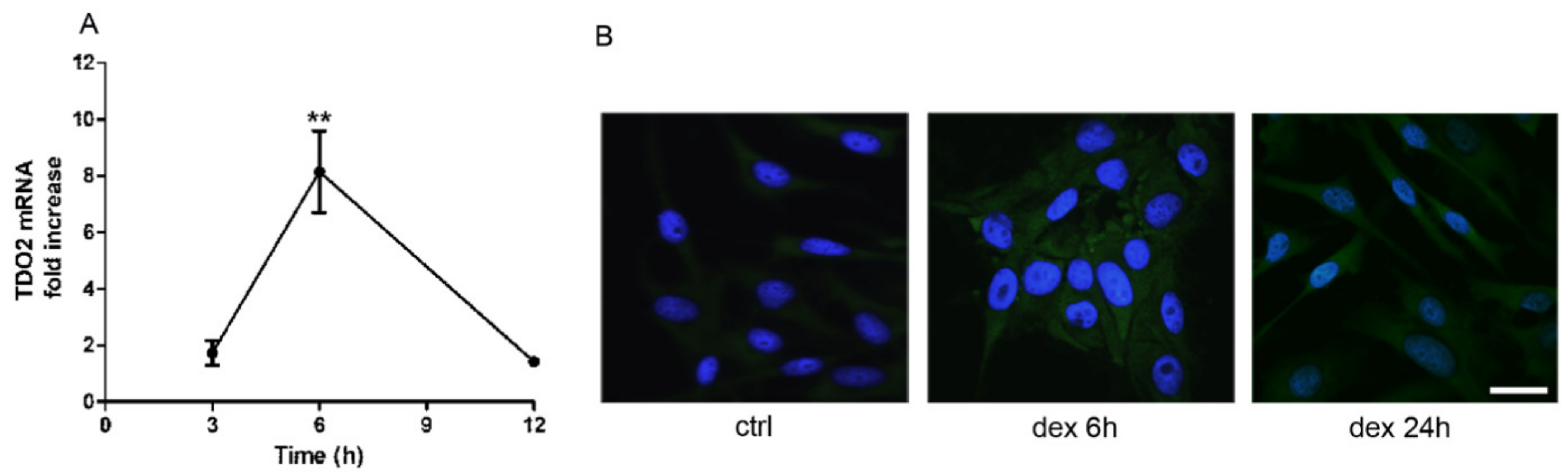
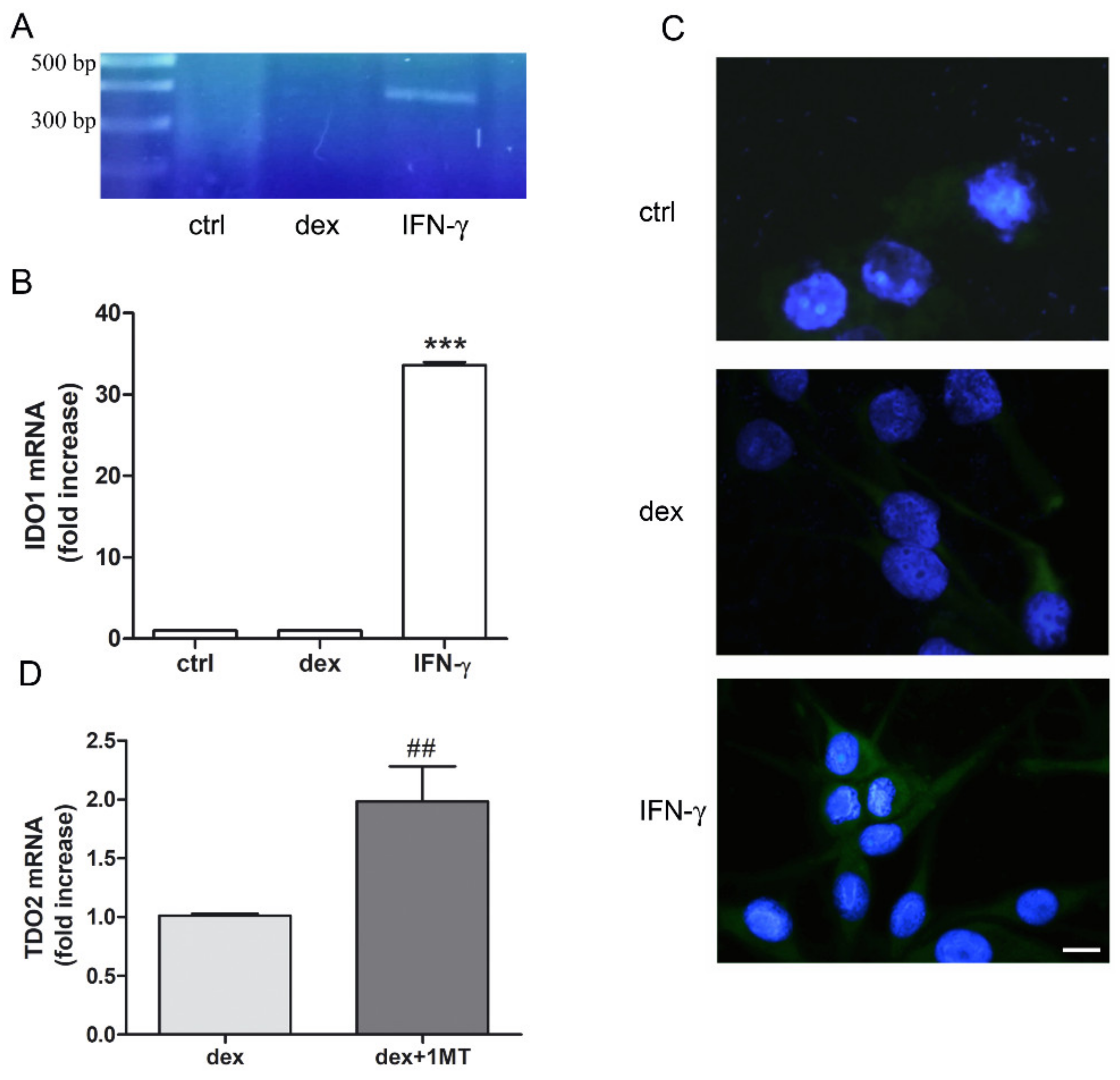
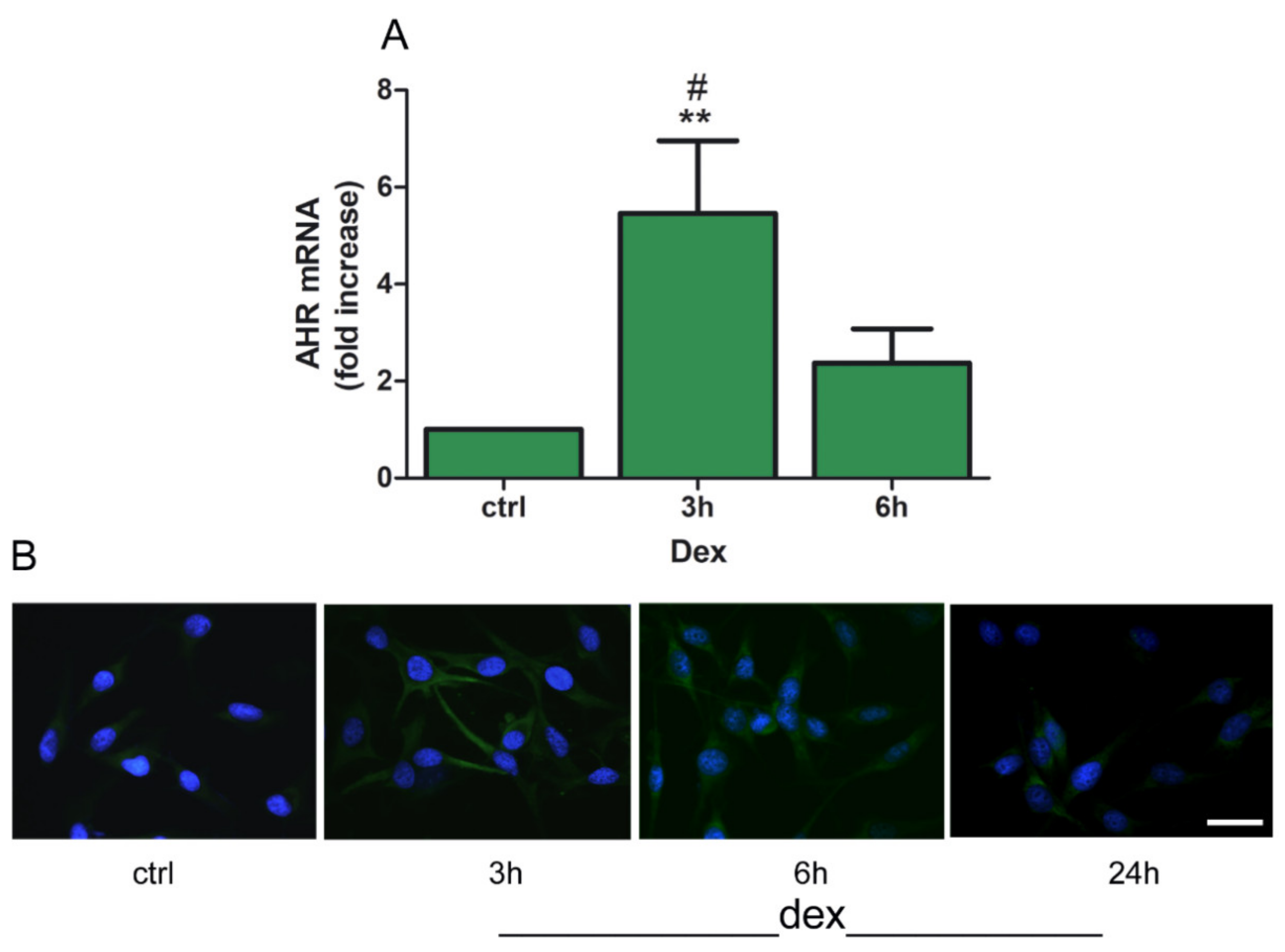
2.1. Dexamethasone Increased TDO and AHR Expression
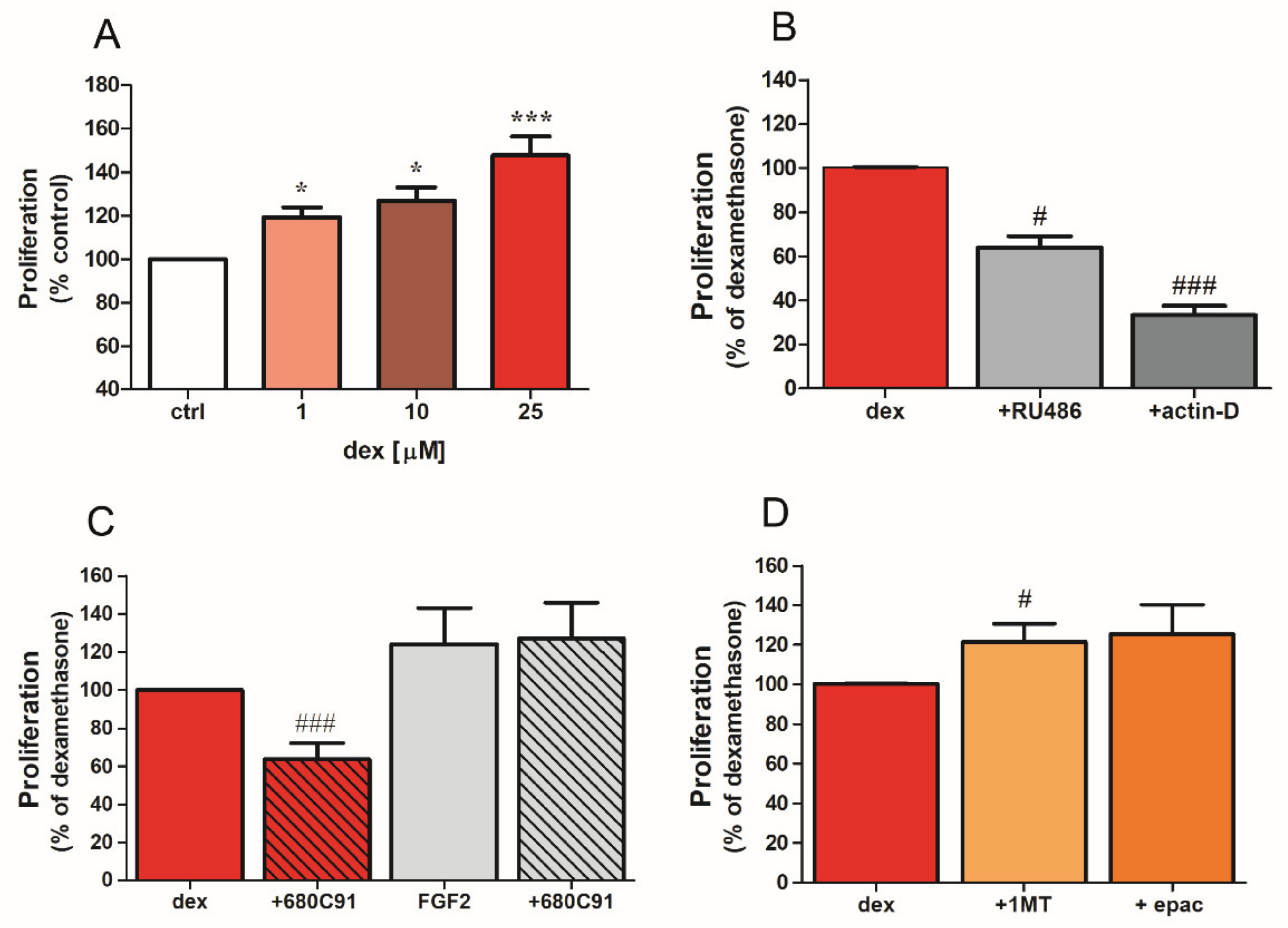
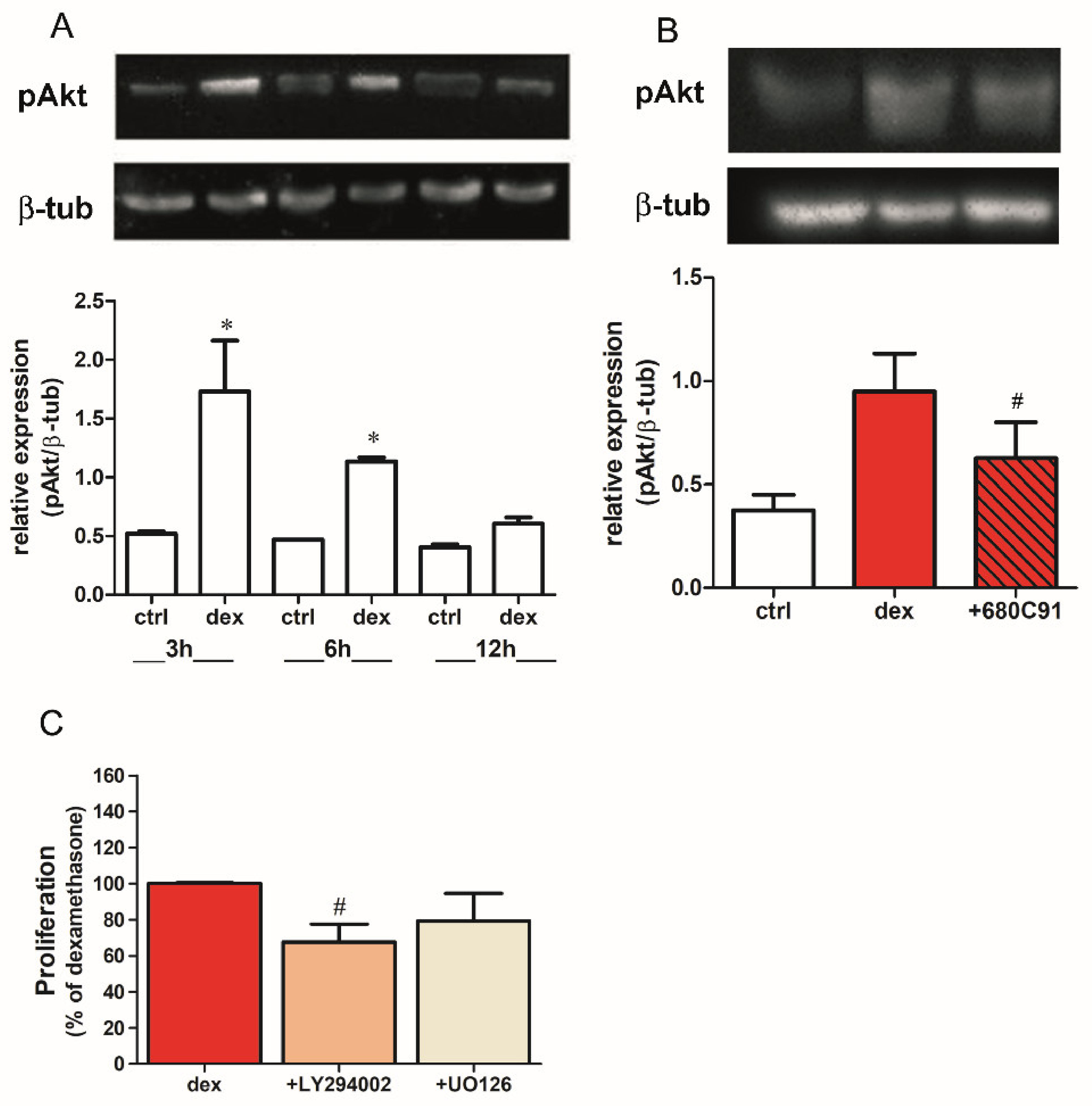
2.2. Dexamethasone Stimulates SK-Mel-28 Proliferation via TDO and PI3K/Akt
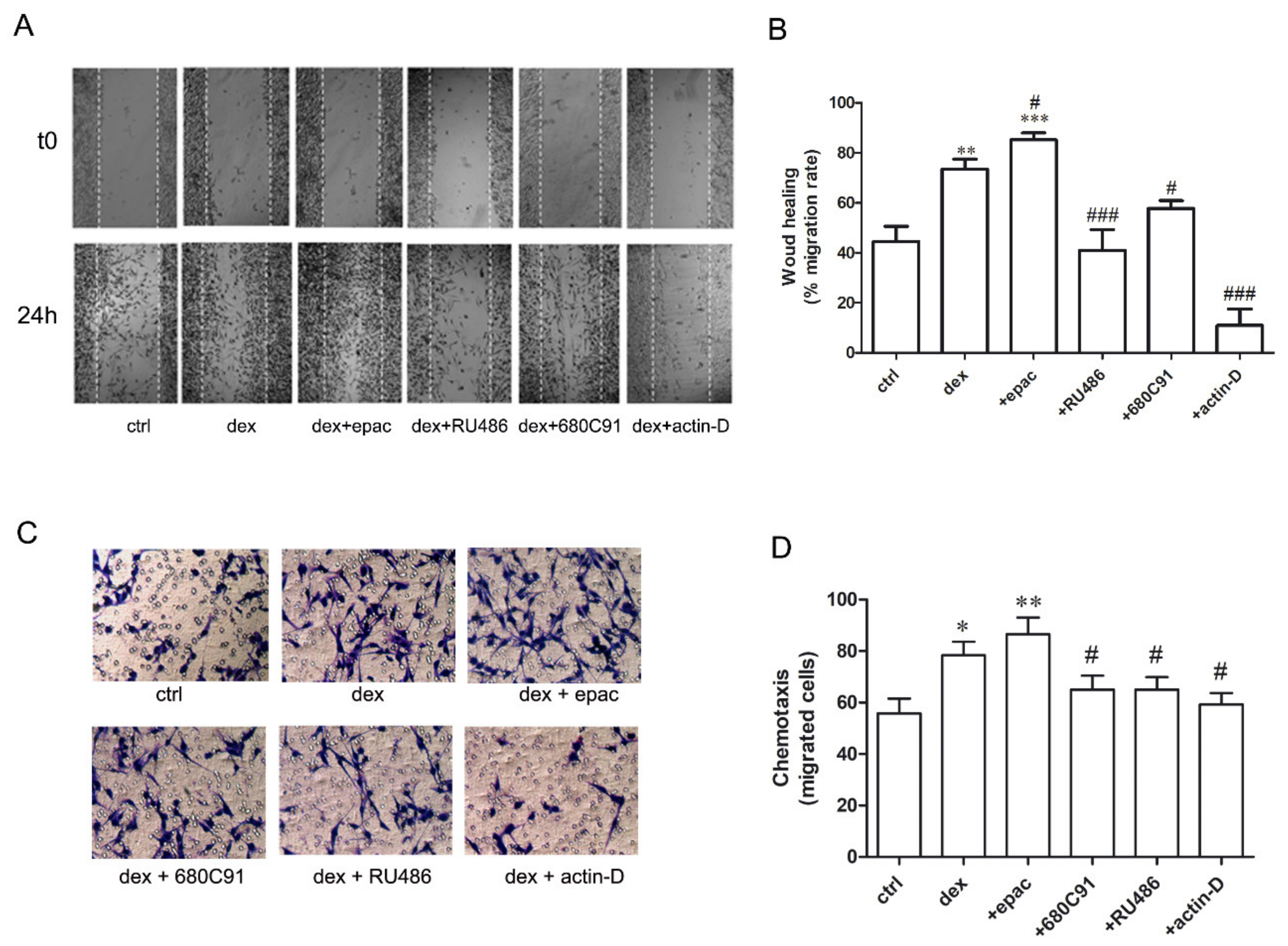
2.3. Dexamethasone Stimulates SK-Mel-28 Migration
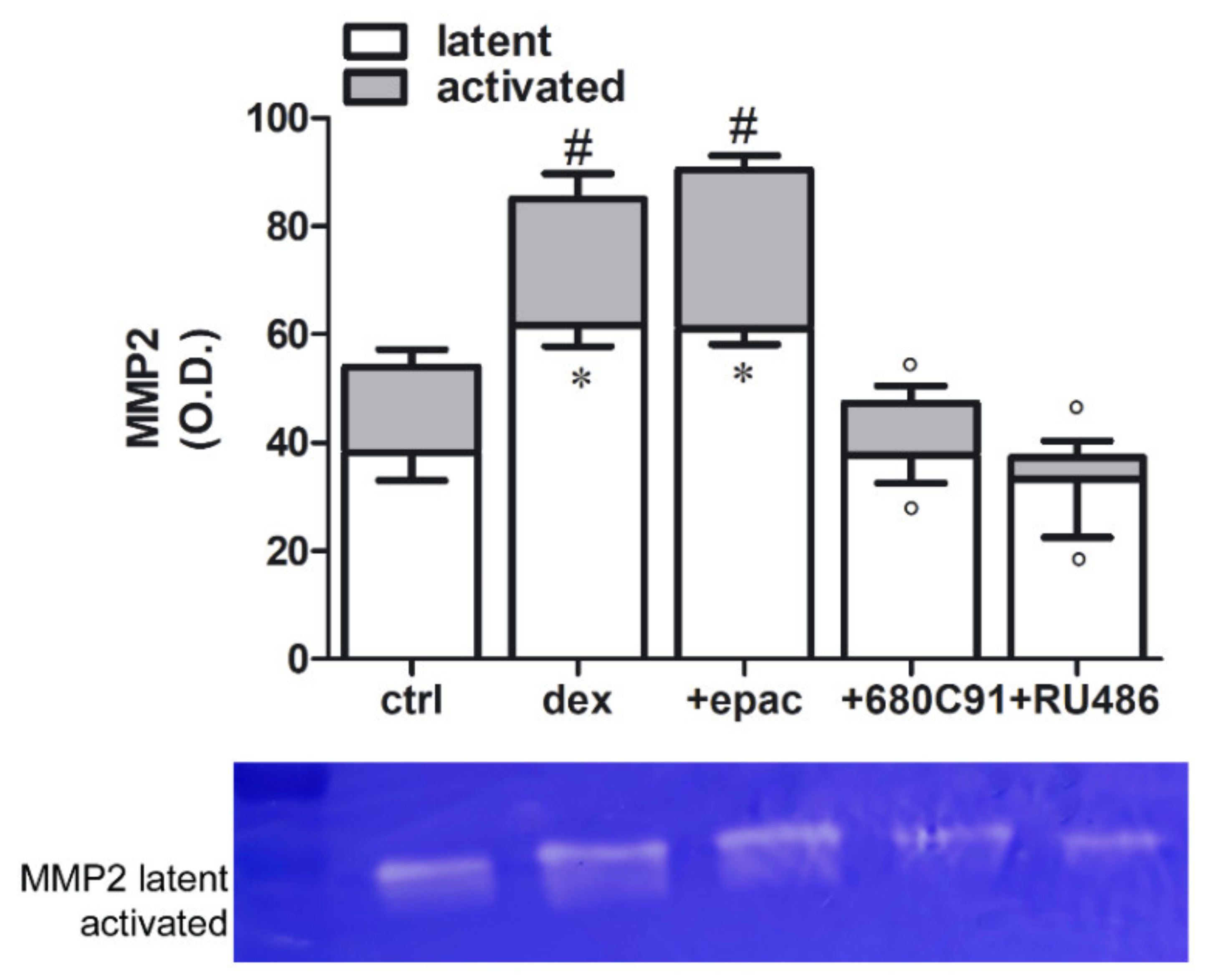
2.4. Dexamethasone Effect on MMP2 Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RT-PCR and Real Time PCR
4.3. Cell Proliferation
4.4. Chemotactic Assay
4.5. Wound-Healing Assay
4.6. Western Blotting Analysis
4.7. Immunofluorescence
4.8. Gelatin Zymography
4.9. Materials
4.10. Statistical Evaluation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Badawy, A.A.-B. Tryptophan Metabolism: A Versatile Area Providing Multiple Targets for Pharmacological Intervention. Egypt. J. Basic Clin. Pharmacol. 2019, 9, 1–48. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Weinlich, G.; Murr, C.; Richardsen, L.; Winkler, C.; Fuchs, D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology 2006, 214, 8–14. [Google Scholar] [CrossRef]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolušić, E.; Frédérick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Kozlova, A.; Frédérick, R. Current state on tryptophan 2,3-dioxygenase inhibitors: A patent review. Expert Opin. Ther. Pat. 2019, 29, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Britan, A.; Maffre, V.; Tone, S.; Drevet, J.R. Quantitative and spatial differences in the expression of tryptophan-metabolizing enzymes in mouse epididymis. Cell Tissue Res. 2006, 342, 301–310. [Google Scholar] [CrossRef]
- Minatogawa, Y.; Suzuki, S.; Ando, Y.; Tone, S.; Takikawa, O. Tryptophan pyrrole ring cleavage enzymes in placenta. Adv Exp Med Biol. 2003, 527, 425–434. [Google Scholar] [PubMed]
- Haber, R.; Bessette, D.; Hulihan-Giblin, B.; Durcan, M.J.; Goldman, D. Identification of Tryptophan 2,3-Dioxygenase RNA in Rodent Brain. J. Neurochem. 1993, 60, 1159–1162. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Somarribas Patterson, L.F.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chiang, S.Y.; Jian, S.F.; Wu, C.Y.; Lin, Y.S.; Tsai, Y.M.; Chou, S.H.; Tsai, M.J.; Kuo, P.L. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 2016, 7, 27584–27598. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.; Jackson, K.C. Role of corticosteroids in palliative care. J. Pain Palliat. Care Pharmacother. 2007, 21, 69–76. [Google Scholar] [CrossRef]
- Cook, A.M.; McDonnell, A.M.; Lake, R.A.; Nowak, A.K. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology 2016, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Beckermann, B.; Kallifatidis, G.; Liu, Z.; Rittgen, W.; Edler, L.; Büchler, P.; Debatin, K.M.; Büchler, M.W.; Friess, H.; et al. Corticosteroids induce chemotherapy resistance in the majority of tumour cells from bone, brain, breast, cervix, melanoma and neuroblastoma. Int. J. Oncol. 2006, 29, 1295–1301. [Google Scholar] [CrossRef] [Green Version]
- Gündisch, S.; Boeckeler, E.; Behrends, U.; Amtmann, E.; Ehrhardt, H.; Jeremias, I. Glucocorticoids augment survival and proliferation of tumor cells. Anticancer Res. 2012, 32, 4251–4262. [Google Scholar]
- Dobos, J.; Kenessey, I.; Tímár, J.; Ladányi, A. Glucocorticoid receptor expression and antiproliferative effect of dexamethasone on human melanoma cells. Pathol. Oncol. Res. 2011, 17, 729–734. [Google Scholar] [CrossRef]
- Comings, D.E.; Muhleman, D.; Dietz, G.; Sherman, M.; Forest, G.L. Sequence of human tryptophan 2, 3-dioxygenase (TDO2): Presence of a glucocorticoid response-like element composed of a GTT repeat and an intronic CCCCT repeat. Genomics 1995, 29, 390–396. [Google Scholar] [CrossRef]
- Paccosi, S.; Cecchi, M.; Silvano, A.; Fabbri, S.; Parenti, A. Different effects of tryptophan 2,3-dioxygenase inhibition on SK-Mel-28 and HCT-8 cancer cell lines. J. Cancer Res. Clin. Oncol. 2020, 146, 3155–3163. [Google Scholar] [CrossRef]
- Stejskalova, L.; Rulcova, A.; Vrzal, R.; Dvorak, Z.; Pavek, P. Dexamethasone accelerates degradation of aryl hydrocarbon receptor (AHR) and suppresses CYP1A1 induction in placental JEG-3 cell line. Toxicol. Lett. 2013, 223, 183–191. [Google Scholar] [CrossRef]
- Cato, A.C.B.; Nestl, A.; Mink, S. Rapid actions of steroid receptors in cellular signaling pathways. Sci. STKE 2002, 2002, re9. [Google Scholar] [CrossRef]
- Limbourg, F.P.; Liao, J.K. Nontranscriptional actions of the glucocorticoid receptor. J. Mol. Med. 2003, 81, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Cordella, M.; Tabolacci, C.; Nassa, G.; D’Arcangelo, D.; Senatore, C.; Pagnotto, P.; Magliozzi, R.; Salvati, A.; Weisz, A.; et al. TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J. Exp. Clin. Cancer Res. 2018, 37, 1–17. [Google Scholar] [CrossRef]
- Pantouris, G.; Mowat, C.G. Antitumour agents as inhibitors of tryptophan 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2014, 443, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.D. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 2008, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wenger, T.; Mattern, J.; Ilea, S.; Frey, C.; Gutwein, P.; Altevogt, P.; Bodenmüller, W.; Gassler, N.; Schnabel, P.A.; et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer Biol. Ther. 2007, 6, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valles, S.L.; Benlloch, M.; Rodriguez, M.L.; Mena, S.; Pellicer, J.A.; Asensi, M.; Obrador, E.; Estrela, J.M. Stress hormones promote growth of B16-F10 melanoma metastases: An interleukin 6- and glutathione-dependent mechanism. J. Transl. Med. 2013, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, D.C.; Deutsch, G.B.; Kirchoff, D.D.; Lee, J.; Huynh, K.T.; Lee, D.Y.; Foshag, L.J.; Bilchik, A.J.; Faries, M.B. Adrenalectomy for metastatic melanoma: Current role in the age of nonsurgical treatments. Am. Surg. 2015, 81, 1005–1009. [Google Scholar] [CrossRef]
- Shimba, S.; Komiyama, K.; Moro, I.; Tezuka, M. Overexpression of the aryl hydrocarbon receptor (AhR) accelerates the cell proliferation of A549 cells. J. Biochem. 2002, 132, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Luecke, S.; Backlund, M.; Jux, B.; Esser, C.; Krutmann, J.; Rannug, A. The aryl hydrocarbon receptor (AHR), a novel regulator of human melanogenesis. Pigment Cell Melanoma Res. 2010, 23, 828–833. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X.; et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2- human breast cancer cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, P.K.; Das Gupta, T.K.; Beattie, C.W.; Walker, M.J. Glucocorticoid-induced exacerbation of metastatic human melanoma. J. Surg. Oncol. 1982, 20, 49–52. [Google Scholar] [CrossRef]
- Huang, G.-X.; Wang, Y.; Su, J.; Zhou, P.; Li, B.; Yin, L.-J.; Lu, J. Up-regulation of Rho-associated kinase 1/2 by glucocorticoids promotes migration, invasion and metastasis of melanoma. Cancer Lett. 2017, 410, 1–11. [Google Scholar] [CrossRef]
- Huang, G.X.; Qi, M.F.; Li, X.L.; Tang, F.; Zhu, L. Involvement of upregulation of fibronectin in the pro-adhesive and pro-survival effects of glucocorticoid on melanoma cells. Mol. Med. Rep. 2018, 17, 3380–3387. [Google Scholar] [CrossRef] [Green Version]
- Krasil’nikov, M.A.; Luzai, E.V.; Scherbakov, A.M.; Shatskaya, V.A.; Shtil, A.A.; Gershtein, E.S. Role of phosphatidylinositol-3 kinase in regulation of differential sensitivity of melanoma cells to antitumor agents. A model for hormone resistance development in tumor cells. Biochemistry 2004, 69, 322–330. [Google Scholar] [CrossRef]
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; De Plaen, E.; Van Den Eynde, B.J. Constitutive IDO1 expression in human tumors is driven by cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol. Res. 2017, 5, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Yang, A.; Ma, J.; Wu, M.; Xu, H.; Wu, K.; Jin, Y.; Xie, Y. Akt2 mediates glucocorticoid resistance in lymphoid malignancies through FoxO3a/Bim axis and serves as a direct target for resistance reversal. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumikawa, T.; Shigeoka, Y.; Igishi, T.; Suyama, H.; Yamasaki, A.; Hashimoto, K.; Matsumoto, S.; Takeda, K.; Ueda, Y.; Shimizu, E. Dexamethasone interferes with trastuzumab-induced cell growth inhibition through restoration of AKT activity in BT-474 breast cancer cells. Int. J. Oncol. 2008, 32, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.X.; Wang, Y.; Fu, C.C.; Diao, F.; Song, L.N.; Li, Z.-B.; Yang, R.; Lu, J. Dexamethasone enhances cell resistance to chemotherapy by increasing adhesion to extracellular matrix in human ovarian cancer cells. Endocr. Relat. Cancer 2010, 17, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Stringer-Reasor, E.M.; Baker, G.M.; Skor, M.N.; Kocherginsky, M.; Lengyel, E.; Fleming, G.F.; Conzen, S.D. Glucocorticoid receptor activation inhibits chemotherapy-induced cell death in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2015, 138, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indini, A.; Rijavec, E.; Grossi, F. Immune related adverse events and response to immunotherapy: Focus on corticosteroids. Lung Cancer 2020, 145, 225. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Kicinski, M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Khattak, A.; Carlino, M.S.; et al. Association between Immune-Related Adverse Events and Recurrence-Free Survival among Patients with Stage III Melanoma Randomized to Receive Pembrolizumab or Placebo: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2020, 6, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Arbour, K.C.; Mezquita, L.; Long, N.; Rizvi, H.; Auclin, E.; Ni, A.; Martínez-Bernal, G.; Ferrara, R.; Victoria Lai, W.; Hendriks, L.E.L.; et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non–small-cell lung cancer. J. Clin. Oncol. 2018, 36, 2872–2878. [Google Scholar] [CrossRef]
- Scott, S.C.; Pennell, N.A. Early Use of Systemic Corticosteroids in Patients with Advanced NSCLC Treated with Nivolumab. J. Thorac. Oncol. 2018, 13, 1771–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohmann, U.; Volpi, C.; Fallarino, F.; Bozza, S.; Bianchi, R.; Vacca, C.; Orabona, C.; Belladonna, M.L.; Ayroldi, E.; Nocentini, G.; et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat. Med. 2007, 13, 579–586. [Google Scholar] [CrossRef]
- Tone, M.; Tone, Y.; Adams, E.; Yates, S.F.; Frewin, M.R.; Cobbold, S.P.; Waldmann, H. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15059–15064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, K.M.; Carlino, M.; Joerger, M.; Di Nicola, M.; Meniawy, T.; Rottey, S.; Moreno, V.; Gazzah, A.; Delord, J.P.; Paz-Ares, L.; et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination with Nivolumab for Patients with Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2020, 6, 100–107. [Google Scholar]
- Paccosi, S.; Musilli, C.; Mangano, G.; Guglielmotti, A.; Parenti, A. The monocyte chemotactic protein synthesis inhibitor bindarit prevents mesangial cell proliferation and extracellular matrix remodeling. Pharmacol. Res. 2012, 66, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Cinci, L.; Luceri, C.; Bigagli, E.; Carboni, I.; Paccosi, S.; Parenti, A.; Guasti, D.; Coronnello, M. Development and characterization of an in vitro model of colorectal adenocarcinoma with MDR phenotype. Cancer Med. 2016, 5, 1279–1291. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cecchi, M.; Paccosi, S.; Silvano, A.; Eid, A.H.; Parenti, A. Dexamethasone Induces the Expression and Function of Tryptophan-2-3-Dioxygenase in SK-MEL-28 Melanoma Cells. Pharmaceuticals 2021, 14, 211. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030211
Cecchi M, Paccosi S, Silvano A, Eid AH, Parenti A. Dexamethasone Induces the Expression and Function of Tryptophan-2-3-Dioxygenase in SK-MEL-28 Melanoma Cells. Pharmaceuticals. 2021; 14(3):211. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030211
Chicago/Turabian StyleCecchi, Marta, Sara Paccosi, Angela Silvano, Ali Hussein Eid, and Astrid Parenti. 2021. "Dexamethasone Induces the Expression and Function of Tryptophan-2-3-Dioxygenase in SK-MEL-28 Melanoma Cells" Pharmaceuticals 14, no. 3: 211. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14030211