
Design and Synthesis of 2,6-Disubstituted-4′-Selenoadenosine-5′-N,N-Dimethyluronamide Derivatives as Human A3 Adenosine Receptor Antagonists



Abstract
:
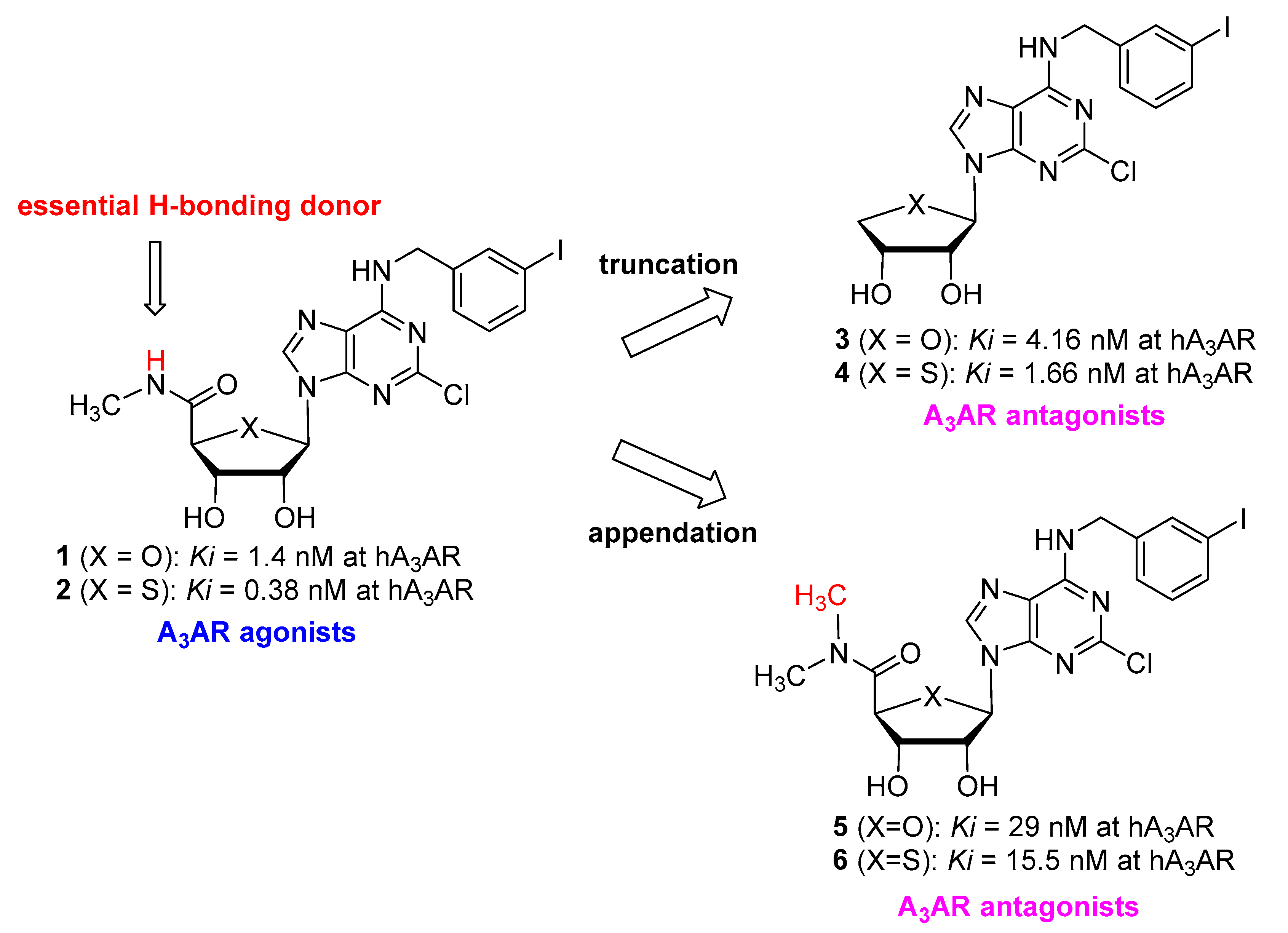
1. Introduction
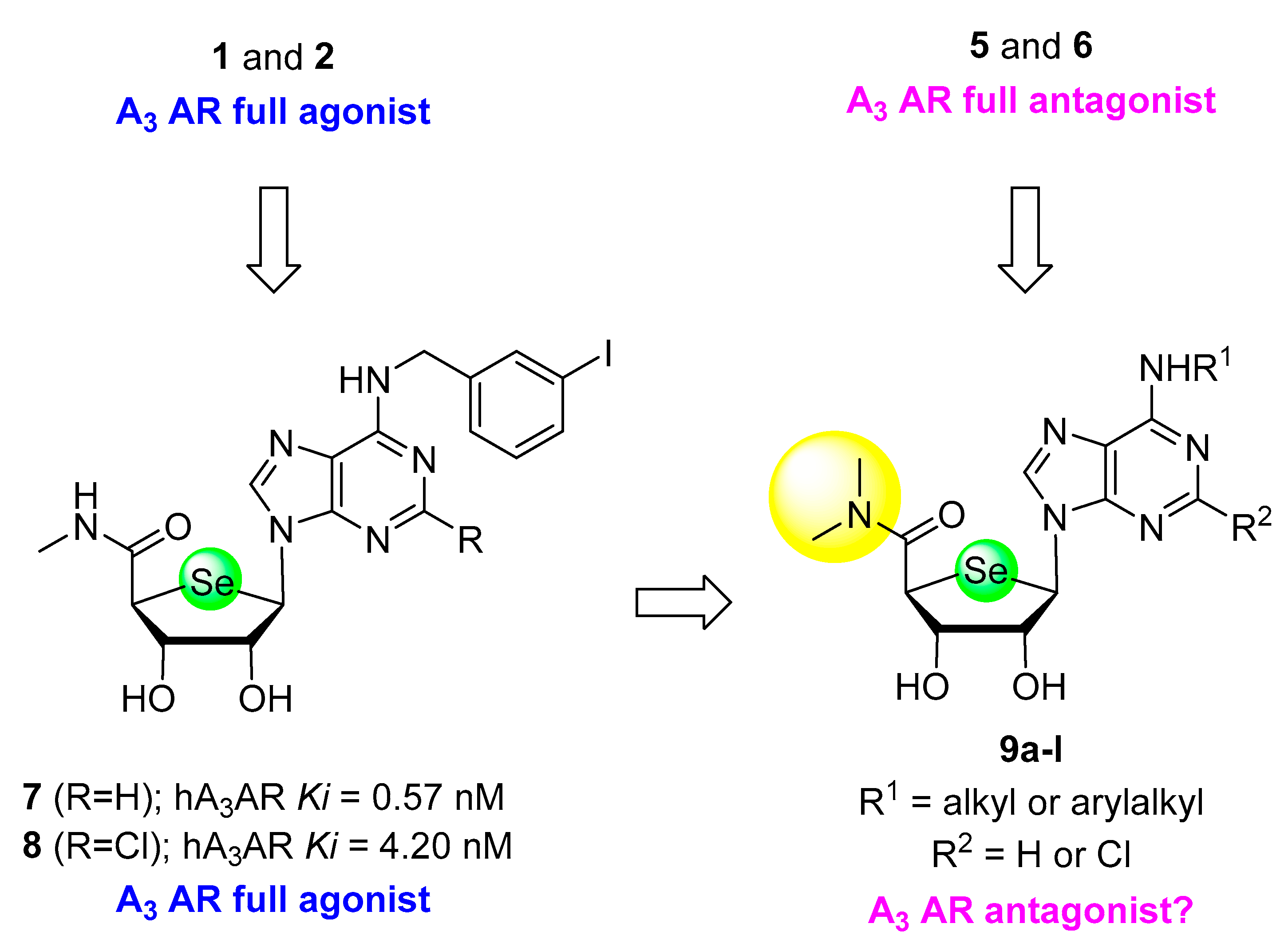
2. Results
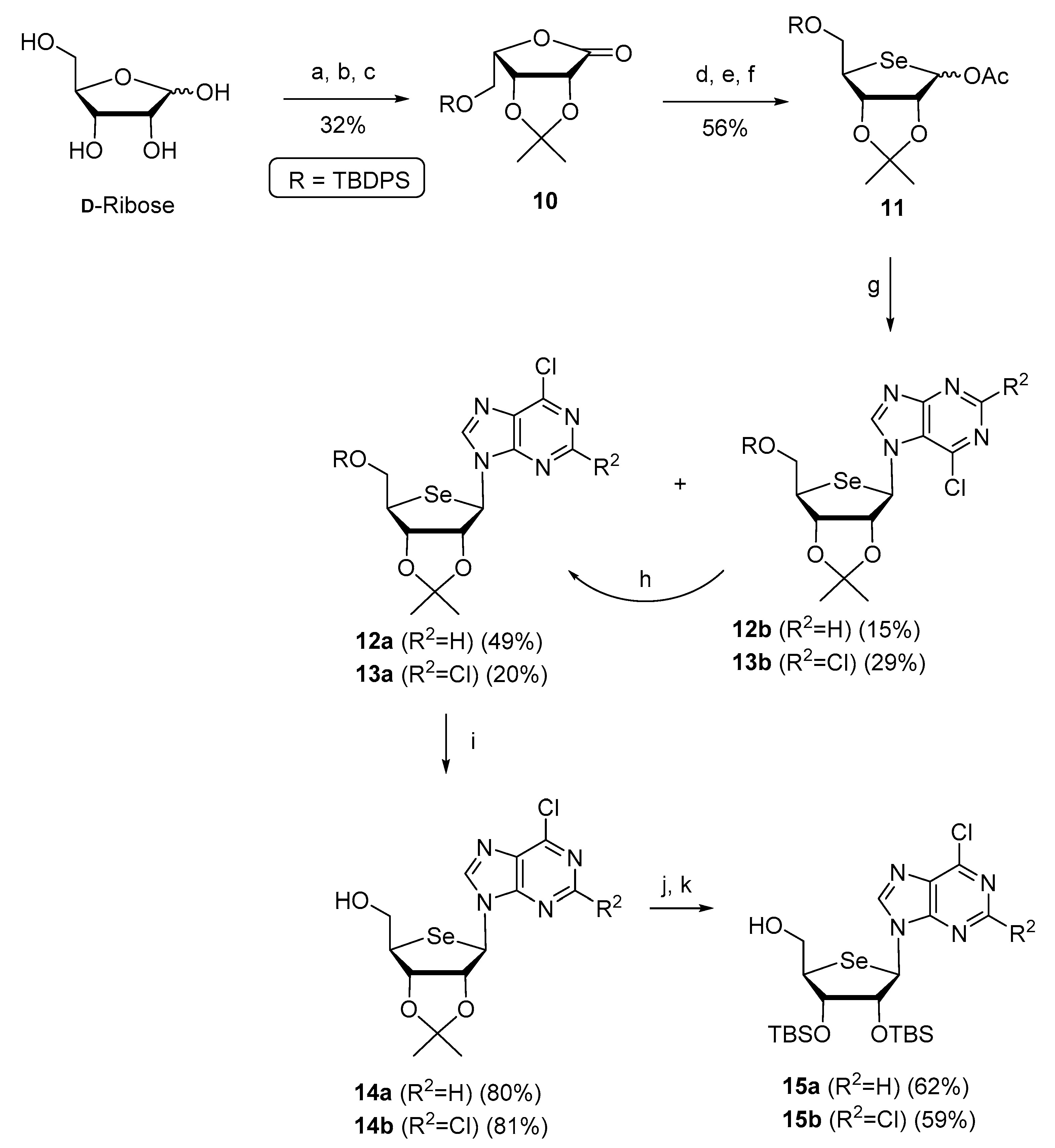
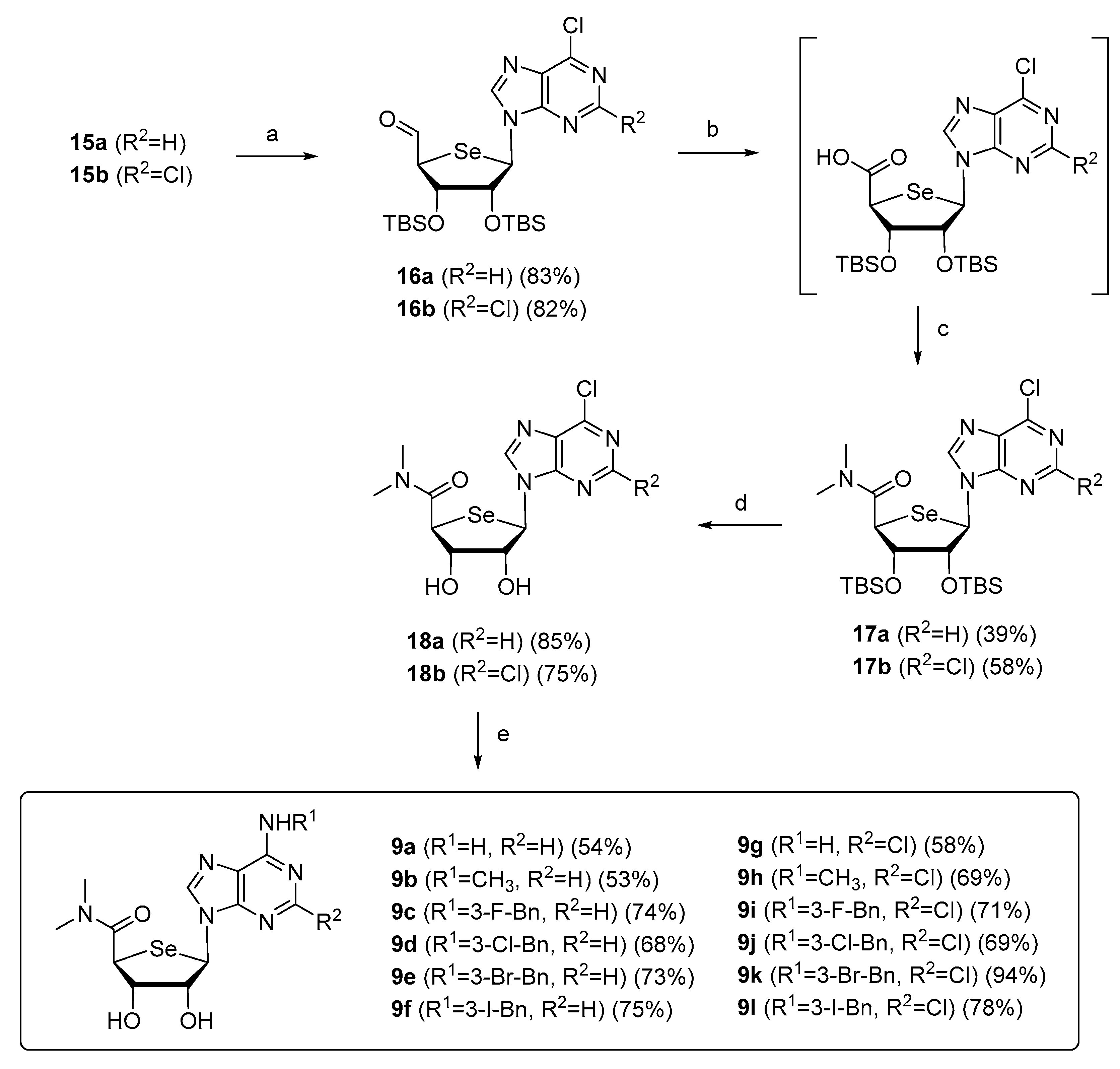
2.1. Chemistry
2.2. Biology
2.2.1. Binding Affinity
2.2.2. CAMP Functional Assay
2.3. Molecular Modelling Studies
3. Materials and Methods
3.1. Chemical Synthesis
General Procedure for the Synthesis of 9a–l
3.2. Biological Evaluation
3.2.1. Binding Assay at hA1AR
3.2.2. Binding Assay at hA2AAR
3.2.3. Binding Assay at hA2BAR
3.2.4. Binding Assay at hA3AR
3.2.5. Cyclic AMP Accumulation Assay
3.3. Molecular Modelling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Varani, K.; Gessi, S.; Merighi, S.; Borea, P.A. Agonists and Antagonists: Molecular Mechanisms and Therapeutic Applications. In A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics; Borea, P., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 301–317. [Google Scholar] [CrossRef]
- Ge, Z.D.; Peart, J.N.; Kreckler, L.M.; Wan, T.C.; Jacobson, M.A.; Gross, G.J.; Auchampach, J.A. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J. Pharmacol. Exp. Ther. 2006, 319, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, D. Purinergic Regulation of Neutrophil Function. Front. Immunol. 2018, 9, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marucci, G.; Santinelli, C.; Buccioni, M.; Navia, A.M.; Lambertucci, C.; Zhurina, A.; Yli-Harja, O.; Volpini, R.; Kandhavelu, M. Anticancer activity study of A3 adenosine receptor agonists. Life Sci. 2018, 205, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011, 226, 2438–2447. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Tabrizi, M.A.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 adenosine receptors as modulators of inflammation: From medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Von Lubitz, D.K.J.E.; Carter, M.F.; Deutsch, S.I.; Lin, R.C.S.; Mastropaolo, J.; Meshulam, Y.; Jacobson, K.A. The effects of adenosine A3 receptor stimulation on seizures in mice. Eur. J. Pharmacol. 1995, 275, 23–29. [Google Scholar] [CrossRef]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Borea, P.A. Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem. Pharmacol. 2006, 72, 19–31. [Google Scholar] [CrossRef]
- Fishman, P.; Cohen, S.; Bar-Yehuda, S. Targeting the A3 adenosine receptor for glaucoma treatment (Review). Mol. Med. Rep. 2013, 7, 1723–1725. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.O.; Ji, X.; Siddiqi, S.M.; Olah, M.E.; Stiles, G.L.; Jacobson, K.A. 2-Substitution of N6-Benzyladenosine-5′-uronamides Enhances Selectivity for A3 Adenosine Receptors. J. Med. Chem. 1994, 37, 3614–3621. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Kim, S.-K.; Biadatti, T.; Chen, W.; Lee, K.; Barak, D.; Kim, S.G.; Johnson, C.R.; Jacobson, K.A. Structural Determinants of A3 Adenosine Receptor Activation: Nucleoside Ligands at the Agonist/Antagonist Boundary. J. Med. Chem. 2002, 45, 4471–4484. [Google Scholar] [CrossRef] [PubMed]
- Jeong, L.S.; Pal, S.; Choe, S.A.; Choi, W.J.; Jacobson, K.A.; Gao, Z.-G.; Klutz, A.M.; Hou, X.; Kim, H.O.; Lee, H.W.; et al. Structure−Activity Relationships of Truncated d- and l-4′-Thioadenosine Derivatives as Species-Independent A3 Adenosine Receptor Antagonists. J. Med. Chem. 2008, 51, 6609–6613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosh, D.K.; Salmaso, V.; Rao, H.; Bitant, A.; Fisher, C.L.; Lieberman, D.I.; Vorbrüggen, H.; Reitman, M.L.; Gavrilova, O.; Gao, Z.-G.; et al. Truncated (N)-Methanocarba Nucleosides as Partial Agonists at Mouse and Human A3 Adenosine Receptors: Affinity Enhancement by N6-(2-Phenylethyl) Substitution. J. Med. Chem. 2020, 63, 4334–4348. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Joshi, B.V.; Klutz, A.M.; Kim, S.-K.; Lee, H.W.; Kim, H.O.; Jeong, L.S.; Jacobson, K.A. Conversion of A3 adenosine receptor agonists into selective antagonists by modification of the 5′-ribofuran-uronamide moiety. Bioorg. Med. Chem. Lett. 2006, 16, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Jeong, L.S.; Lee, H.W.; Kim, H.O.; Tosh, D.K.; Pal, S.; Choi, W.J.; Gao, Z.G.; Patel, A.R.; Williams, W.; Jacobson, K.A.; et al. Structure-activity relationships of 2-chloro-N6-substituted-4′-thioadenosine-5′-N,N-dialkyluronamides as human A3 adenosine receptor antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 1612–1616. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, L.X.; Park, J.; Lee, H.W.; Sahu, P.K.; Cui, M.; Moss, S.M.; Hammes, E.; Warnick, E.; Gao, Z.-G.; et al. N6-Substituted 5′-N-Methylcarbamoyl-4′-selenoadenosines as Potent and Selective A3 Adenosine Receptor Agonists with Unusual Sugar Puckering and Nucleobase Orientation. J. Med. Chem. 2017, 60, 3422–3437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Mannes, P.; Jung, Y.-H.; Ciancetta, A.; Bitant, A.; Lieberman, D.I.; Khaznadar, S.; Auchampach, J.A.; Gao, Z.-G.; Jacobson, K.A. Structure activity relationship of 2-arylalkynyl-adenine derivatives as human A3 adenosine receptor antagonists. Med. Chem. Comm. 2018, 9, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, R.; Torquati, I.; Kachler, S.; Luongo, L.; Maione, S.; Franchetti, P.; Grifantini, M.; Novellino, E.; Lavecchia, A.; Klotz, K.-N.; et al. 5′-C-Ethyl-tetrazolyl-N6-Substituted Adenosine and 2-Chloro-adenosine Derivatives as Highly Potent Dual Acting A1 Adenosine Receptor Agonists and A3 Adenosine Receptor Antagonists. J. Med. Chem. 2015, 58, 2560–2566. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Affinity, Ki, nM ± SEM a,b (or % Inhibition at 10 uM) | |||||||
| X | Y | R1 | R2 | hA1AR | hA2AAR | hA2BAR | hA3AR | |
| 5 c | O | Cl | 3-I-Bn | CH3 | 5870 ± 930 | >10,000 | >10,000 | 29.0 ± 4.9 |
| 6 c | S | Cl | 3-I-Bn | CH3 | 6220 ± 640 | >10,000 | >10,000 | 15.5 ± 3.1 |
| 7 d | Se | H | 3-I-Bn | H | 480 ± 94 | 1080 ± 140 | ND | 0.57 ± 0.10 |
| 8 d | Se | Cl | 3-I-Bn | H | 311 ± 47 | 1200 ± 70 | ND | 4.20 ± 0.73 |
| 9a | Se | H | H | CH3 | 16% ± 4 | 22% ± 3 | 17% ± 1 | 3710 ± 600 |
| 9b | Se | H | CH3 | CH3 | 9% ± 2 | 3% ± 2 | 9% ± 4 | 609 ± 47 |
| 9c | Se | H | 3-F-Bn | CH3 | 13% ± 2 | 12% ± 1 | 19% ± 5 | 2020 ± 170 |
| 9d | Se | H | 3-Cl-Bn | CH3 | 23% ± 5 | 4% ± 3 | 15% ± 1 | 1190 ± 160 |
| 9e | Se | H | 3-Br-Bn | CH3 | 43% ± 7 | 37% ± 8 | ND | 36.3 ± 12.7 |
| 9f | Se | H | 3-I-Bn | CH3 | 36% ± 6 | 32% ± 1 | ND | 22.7 ± 8.9 |
| 9g | Se | Cl | H | CH3 | 9% ± 1 | 3% ± 3 | 25% ± 5 | 3250 ± 370 |
| 9h | Se | Cl | CH3 | CH3 | 58% ± 7 | 5% ± 2 | 19% ± 5 | 1060 ± 140 |
| 9i | Se | Cl | 3-F-Bn | CH3 | 34% ± 2 | 9% ± 4 | 14% ± 2 | 238 ± 37 |
| 9j | Se | Cl | 3-Cl-Bn | CH3 | 63% ± 5 | 13% ± 1 | 27% ± 4 | 195 ± 40 |
| 9k | Se | Cl | 3-Br-Bn | CH3 | 58% ± 2 | 10% ± 4 | 27% ± 6 | 180 ± 12 |
| 9l | Se | Cl | 3-I-Bn | CH3 | 68% ± 2 | 10% ± 1 | 23% ± 3 | 253 ± 29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Jacobson, K.A.; Yu, J.; Jeong, L.S. Design and Synthesis of 2,6-Disubstituted-4′-Selenoadenosine-5′-N,N-Dimethyluronamide Derivatives as Human A3 Adenosine Receptor Antagonists. Pharmaceuticals 2021, 14, 363. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14040363
Choi H, Jacobson KA, Yu J, Jeong LS. Design and Synthesis of 2,6-Disubstituted-4′-Selenoadenosine-5′-N,N-Dimethyluronamide Derivatives as Human A3 Adenosine Receptor Antagonists. Pharmaceuticals. 2021; 14(4):363. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14040363
Chicago/Turabian StyleChoi, Hongseok, Kenneth A. Jacobson, Jinha Yu, and Lak Shin Jeong. 2021. "Design and Synthesis of 2,6-Disubstituted-4′-Selenoadenosine-5′-N,N-Dimethyluronamide Derivatives as Human A3 Adenosine Receptor Antagonists" Pharmaceuticals 14, no. 4: 363. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14040363