
miR-34a-Mediated Survivin Inhibition Improves the Antitumor Activity of Selinexor in Triple-Negative Breast Cancer

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
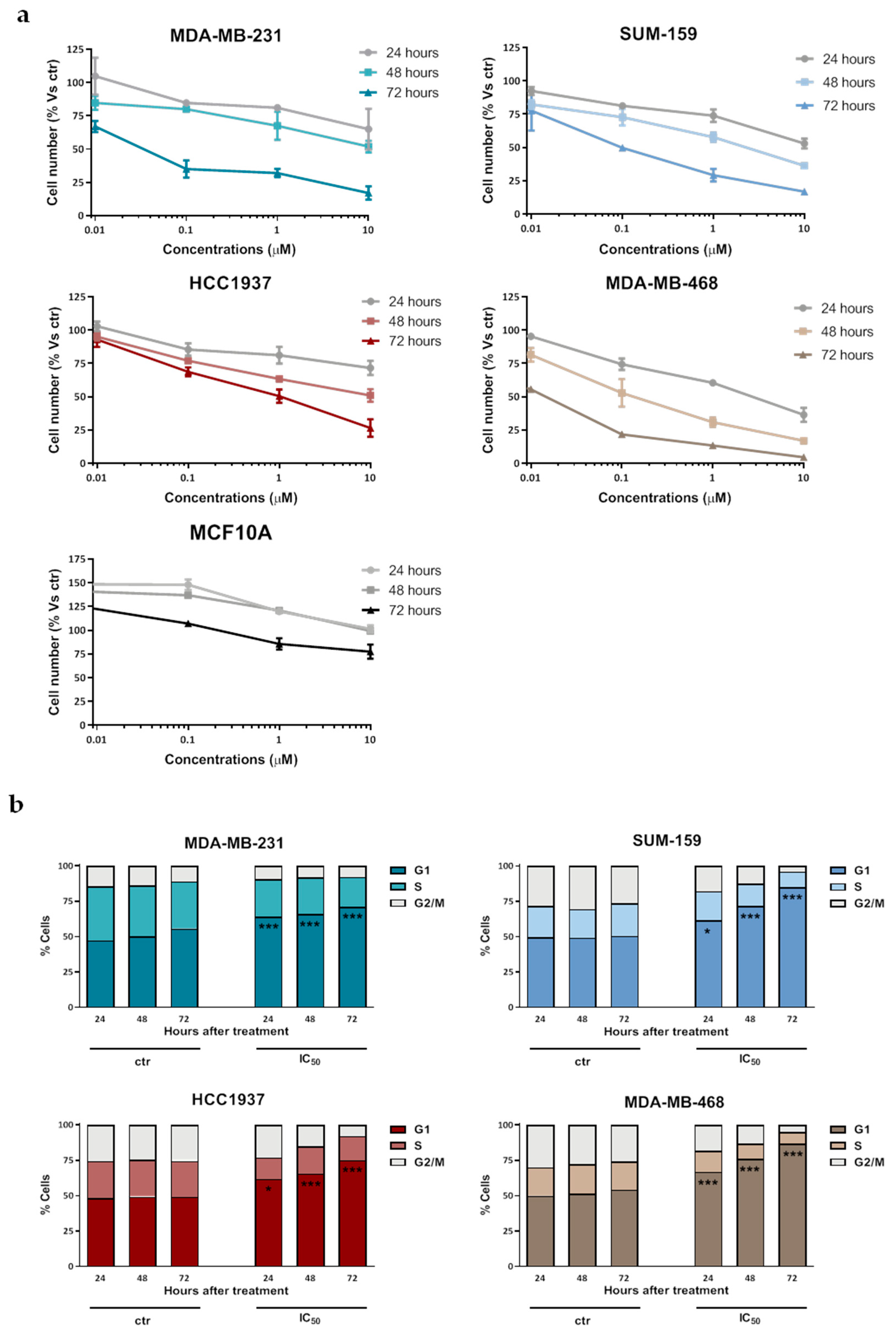
2.1. Selinexor Inhibits TNBC Cell Growth
2.2. Selinexor Impairs Survivin Expression and Intracellular Distribution, and Enhances Apoptosis in TNBC Cells
2.3. Selinexor Variably Affects the Growth of TNBC Xenografts
2.4. miR-34a Ectopic Expression Inhibits Survivin Expression and Induces TNBC Cell Growth Delay
2.5. miR-35a Ectopic Expression Improves the Antiproliferative and Antitumor Activity of Selinexor in TNBC Cells and Xenografts
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Transfection
4.3. Cell Growth Inhibition Assay
4.4. Cell Cycle Distribution Analysis
4.5. Caspase-3 Catalytic Activity
4.6. Protein Extraction and Western Blot Analysis
4.7. ELISA Assay
4.8. Quantitative RT-PCR
4.9. Migration Assay
4.10. In Vivo Experiments
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, M.-C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 2012, 83, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, L.; Sun, Z.; Yao, Y.; Shen, Z.; Wang, H.; Liu, X.; Zhou, F.; Xiang, J.; Yao, R.; Niu, H. CRM1, a novel independent prognostic factor overexpressed in invasive breast carcinoma of poor prognosis. Oncol. Lett. 2018, 15, 7515–7522. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y. Selinexor: First Global Approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Kashyap, T.; Nguyen, T.; Yadav, G.; Chang, H.; Landesman, Y.; Chung, J. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget 2017, 8, 52935–52947. [Google Scholar] [CrossRef] [Green Version]
- Martini, S.; Figini, M.; Croce, A.; Frigerio, B.; Pennati, M.; Gianni, A.M.; De Marco, C.; Daidone, M.G.; Argueta, C.; Landesman, Y.; et al. Selinexor Sensitizes TRAIL-R2-Positive TNBC Cells to the Activity of TRAIL-R2xCD3 Bispecific Antibody. Cells 2020, 9, 2231. [Google Scholar] [CrossRef] [PubMed]
- Arango, N.P.; Yuca, E.; Zhao, M.; Evans, K.W.; Scott, S.; Kim, C.; Gonzalez-Angulo, A.M.; Janku, F.; Ueno, N.T.; Tripathy, D.; et al. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shafique, M.; Ismail-Khan, R.; Extermann, M.; Sullivan, D.; Goodridge, D.; Boulware, D.; Hogue, D.; Soliman, H.; Khong, H.; Han, H.S. A Phase II Trial of Selinexor (KPT-330) for Metastatic Triple-Negative Breast Cancer. Oncologist 2019, 24, 887-e416. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.S.; Musi, E.; Schwartz, G.K. Selinexor (KPT-330) Induces Tumor Suppression through Nuclear Sequestration of IκB and Downregulation of Survivin. Clin. Cancer Res. 2017, 23, 4301–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cesare, M.; Cominetti, D.; Doldi, V.; Lopergolo, A.; Deraco, M.; Gandellini, P.; Friedlander, S.; Landesman, Y.; Kauffman, M.G.; Shacham, S.; et al. Anti-tumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: The role of survivin. Oncotarget 2015, 6, 13119–13132. [Google Scholar] [CrossRef] [Green Version]
- Kazim, S.; Malafa, M.P.; Coppola, D.; Husain, K.; Zibadi, S.; Kashyap, T.; Crochiere, M.; Landesman, Y.; Rashal, T.; Sullivan, D.M.; et al. Selective Nuclear Export Inhibitor KPT-330 Enhances the Antitumor Activity of Gemcitabine in Human Pancreatic Cancer. Mol. Cancer Ther. 2015, 14, 1570–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Mancini, A.; Sanita, P.; Vitale, F.; Marampon, F.; Ventura, L.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin in prostate cancer models. BMC Cancer 2015, 15, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.D.; Wali, V.B.; Cheng, C.J.; Inukai, S.; Booth, C.J.; Agarwal, S.; Rimm, D.L.; Győrffy, B.; Santarpia, L.; Pusztai, L.; et al. miR-34a Silences c-SRC to Attenuate Tumor Growth in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Fan, R.; Wang, L.; Cheng, S.; Li, H.; Jiang, J.; Geng, M.; Jin, Y.; Wu, Y. Expression and regulatory function of miRNA-34a in targeting survivin in gastric cancer cells. Tumor Biol. 2012, 34, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, X.-G.; Hu, X.; Song, T.; Ou, X.; Zhang, C.; Zhang, W.; Zhang, C. MiR-34a and miR-203 Inhibit Survivin Expression to Control Cell Proliferation and Survival in Human Osteosarcoma Cells. J. Cancer 2016, 7, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Geng, D.; Song, X.; Ning, F.; Song, Q.; Yin, H. MiR-34a Inhibits Viability and Invasion of Human Papillomavirus–Positive Cervical Cancer Cells by Targeting E2F3 and Regulating Survivin. Int. J. Gynecol. Cancer 2015, 25, 707–713. [Google Scholar] [CrossRef]
- Stauber, R.H.; Mann, W.; Knauer, S.K. Nuclear and Cytoplasmic Survivin: Molecular Mechanism, Prognostic, and Therapeutic Potential: Figure 1. Cancer Res. 2007, 67, 5999–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.A.; Span, S.W.; Ferreira, C.G.; Kruyt, F.A.; Giaccone, G. CRM1-Mediated Nuclear Export Determines the Cytoplasmic Localization of the Antiapoptotic Protein Survivin. Exp. Cell Res. 2002, 275, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-S.; Wong, C.H.; Huang, Y.-F.; Li, H.-Y. Survivin withdrawal by nuclear export failure as a physiological switch to commit cells to apoptosis. Cell Death Dis. 2010, 1, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Holloway, M.P.; Nguyen, K.; McCauley, D.; Landesman, Y.; Kauffman, M.G.; Shacham, S.; Altura, R.A. XPO1 (CRM1) Inhibition Represses STAT3 Activation to Drive a Survivin-Dependent Oncogenic Switch in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.; Climent, M.; Panebianco, F.; Tordonato, C.; Santoro, A.; Marzi, M.J.; Pelicci, P.G.; Ventura, A.; Nicassio, F. Dual role for miR-34a in the control of early progenitor proliferation and commitment in the mammary gland and in breast cancer. Oncogene 2019, 38, 360–374. [Google Scholar] [CrossRef]
- Imani, S.; Wei, C.; Cheng, J.; Khan, A.; Fu, S.; Yang, L.; Tania, M.; Zhang, X.; Xiao, X.; Zhang, X.; et al. MicroRNA-34a targets epithelial to mesenchymal transition-inducing transcription factors (EMT-TFs) and inhibits breast cancer cell migration and invasion. Oncotarget 2017, 8, 21362–21379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayraktar, R.; Ivan, C.; Bayraktar, E.; Kanlikilicer, P.; Kabil, N.N.; Kahraman, N.; Mokhlis, H.A.; Karakas, D.; Rodriguez-Aguayo, C.; Arslan, A.; et al. Dual Suppressive Effect of miR-34a on the FOXM1/eEF2-Kinase Axis Regulates Triple-Negative Breast Cancer Growth and Invasion. Clin. Cancer Res. 2018, 24, 4225–4241. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.D.; Parsons, C.; Slack, F.J. The tumor-suppressive and potential therapeutic functions of miR-34a in epithelial carcinomas. Expert Opin. Ther. Targets 2016, 20, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Satta, A.; Mezzanzanica, D.; Caroli, F.; Frigerio, B.; Di Nicola, M.; Kontermann, R.E.; Iacovelli, F.; Desideri, A.; Anichini, A.; Canevari, S.; et al. Design, selection and optimization of an anti-TRAIL-R2/anti-CD3 bispecific antibody able to educate T cells to recognize and destroy cancer cells. mAbs 2018, 10, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Satta, A.; Grazia, G.; Caroli, F.; Frigerio, B.; Di Nicola, M.; Raspagliesi, F.; Mezzanzanica, D.; Zaffaroni, N.; Gianni, A.M.; Anichini, A.; et al. A Bispecific Antibody to Link a TRAIL-Based Antitumor Approach to Immunotherapy. Front. Immunol. 2019, 10, 2514. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.N.; Casas, C.G.; Day, E.S. Photoresponsive miR-34a/Nanoshell Conjugates Enable Light-Triggered Gene Regulation to Impair the Function of Triple-Negative Breast Cancer Cells. Nano Lett. 2021, 21, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, C.H.; Ioele, S.A.; Day, E.S. Layer-by-layer assembled PLGA nanoparticles carrying miR-34a cargo inhibit the proliferation and cell cycle progression of triple-negative breast cancer cells. J. Biomed. Mater. Res. Part A 2020, 108, 601–613. [Google Scholar] [CrossRef] [PubMed]
- El Bezawy, R.; De Cesare, M.; Pennati, M.; Deraco, M.; Gandellini, P.; Zuco, V.; Zaffaroni, N. Antitumor activity of miR-34a in peritoneal mesothelioma relies on c-MET and AXL inhibition: Persistent activation of ERK and AKT signaling as a possible cytoprotective mechanism. J. Hematol. Oncol. 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donev, I.S.; Wang, W.; Yamada, T.; Li, Q.; Takeuchi, S.; Matsumoto, K.; Yamori, T.; Nishioka, Y.; Sone, S.; Yano, S. Transient PI3K Inhibition Induces Apoptosis and Overcomes HGF-Mediated Resistance to EGFR-TKIs in EGFR Mutant Lung Cancer. Clin. Cancer Res. 2011, 17, 2260–2269. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Abe, A.; Imagama, S.; Nomura, Y.; Tanizaki, R.; Minami, Y.; Hayakawa, F.; Ito, Y.; Katsumi, A.; Yamamoto, K.; et al. BCR-ABL-independent and RAS / MAPK pathway-dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur. J. Haematol. 2010, 84, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.H.; Morgan, C.R.; Hildebrand-Zanki, S.U. In Vitro Pharmacodynamics of 1-β-D- Arabinofuranosylcytosine: Synergy of Aztitumor Activity with cis-Diamminedichloroplatinum(II). Cancer Res. 1988, 48, 117–121. [Google Scholar] [PubMed]
- Fragomeni, R.A.S.; Chung, H.W.; Landesman, Y.; Senapedis, W.; Saint-Martin, J.-R.; Tsao, H.; Flaherty, K.T.; Shacham, S.; Kauffman, M.; Cusack, J.C. CRM1 and BRAF Inhibition Synergize and Induce Tumor Regression in BRAF-Mutant Melanoma. Mol. Cancer Ther. 2013, 12, 1171–1179. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (µM) 1 | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| MDA-MB-231 | >10 | >10 | 0.05 ± 0.02 |
| SUM-159 | >10 | 3.1 ± 1.1 | 0.12 ± 0.04 |
| HCC1937 | >10 | >10 | 1.04 ± 0.03 |
| MDA-MB-468 | 3.7 ± 1.4 | 0.18 ± 0.06 | 0.03 ± 0.02 |
| Xenografts Model | Treatment (mg/kg) | Schedule | Route | Max TVI% (Day) a |
|---|---|---|---|---|
| MDA-MB-231 | 10 | q3-4d × 8 | p.o. | 25(31) |
| SUM-159 | 10 | q3-4d × 8 | p.o. | 67(28) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, S.; Zuco, V.; Tortoreto, M.; Percio, S.; Campi, E.; El Bezawy, R.; Doldi, V.; Landesman, Y.; Pennati, M.; Zaffaroni, N. miR-34a-Mediated Survivin Inhibition Improves the Antitumor Activity of Selinexor in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 523. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060523
Martini S, Zuco V, Tortoreto M, Percio S, Campi E, El Bezawy R, Doldi V, Landesman Y, Pennati M, Zaffaroni N. miR-34a-Mediated Survivin Inhibition Improves the Antitumor Activity of Selinexor in Triple-Negative Breast Cancer. Pharmaceuticals. 2021; 14(6):523. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060523
Chicago/Turabian StyleMartini, Silvia, Valentina Zuco, Monica Tortoreto, Stefano Percio, Elisa Campi, Rihan El Bezawy, Valentina Doldi, Yosef Landesman, Marzia Pennati, and Nadia Zaffaroni. 2021. "miR-34a-Mediated Survivin Inhibition Improves the Antitumor Activity of Selinexor in Triple-Negative Breast Cancer" Pharmaceuticals 14, no. 6: 523. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060523