
Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP

Phico Therapeutics, Bertarelli Building, Bourn Hall, Bourn, Cambridge CB23 2TN, UK
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2021, 14(10), 1038; https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101038
Submission received: 19 August 2021
/
Revised: 3 October 2021
/
Accepted: 8 October 2021
/
Published: 12 October 2021
(This article belongs to the Special Issue Bacteriophages as Therapeutic Delivery Vehicles)
Abstract
:The difficulties in developing novel classes of antibacterials is leading to a resurgence of interest in bacteriophages as therapeutic agents, and in particular engineered phages that can be optimally designed. Here, pre-clinical microbiology assessment is presented of a Staphylococcus aureus phage engineered to deliver a gene encoding an antibacterial small acid soluble spore protein (SASP) and further, rendered non-lytic to give product SASPject PT1.2. PT1.2 has been developed initially for nasal decolonisation of S. aureus, including methicillin-resistant S. aureus. Time-kill curve assays were conducted with PT1.2 against a range of staphylococcal species, and serial passaging experiments were conducted to investigate the potential for resistance to develop. SASPject PT1.2 demonstrates activity against 100% of 225 geographically diverse S. aureus isolates, exquisite specificity for S. aureus, and a rapid speed of kill. The kinetics of S. aureus/PT1.2 interaction is examined together with demonstrating that PT1.2 activity is unaffected by the presence of human serum albumin. SASPject PT1.2 shows a low propensity for resistance to develop with no consistent shift in sensitivity in S. aureus cells passaged for up to 42 days. SASPject PT1.2 shows promise as a novel first-in-class antibacterial agent and demonstrates potential for the SASPject platform.
1. Introduction
Given that the World Health Organisation (WHO) have stated that antibiotic resistance is one of the biggest threats to global health, new approaches to treating infectious diseases are clearly needed. In recent decades, commercial antibiotic development has frequently involved producing next-generations of existing classes of antibiotics, with novel classes being relatively few and far between [1].
Conventional antibiotics are generally broad spectrum in their activity, being unselective for the specific species of bacteria that is causing a particular infection. Whilst this is clearly valuable in treating patients prior to understanding what the causative pathogen is, it does have an impact on the composition of the microbiota, that is the indigenous microbes found ubiquitously in and on the human body, including in the gut and on skin [2]. Bacteriophages are ubiquitous with an estimated 1031 particles on the planet, and it seems highly probable that every bacterial species or strain will have phage that can infect it [3,4,5]. However, phages can be, and in fact most commonly are, exquisitely selective for their target species or strain of bacteria. This attribute opens up a broad scope for the applicability of phages in very diverse areas, including human and animal health, agriculture, and horticulture. Indeed, as medical understanding of the relevance to health of the microbiota increases, so phages are superbly placed to selectively manipulate the microbiota.
In fact, bacteriophages have a long and chequered history as antibacterial therapeutic agents [6,7,8]. However, as knowledge of bacteriophage biology, specificity, behaviour, and pharmacokinetics has increased, in parallel with genetic tools, so the possibility of engineering phages to optimise or change their characteristics for clinical application has become not just possible, but a reality [3]. This is enabling a new generation of commercially robust opportunities for phage-based antibacterials.
There are now a number of precision phage engineering methods which have been developed, including the Bacteriophage Recombineering with Electroporated DNA (BRED) method [9], the Host Range Determinant Selection (HORDS) method [10], cell-free transcription/translation (TX-TL) systems [3], in vitro assembly of an engineered phage genome from DNA fragments [3], and the use of yeast expression vectors to capture and engineer genomes prior to recovery of phages following transformation of a suitable bacterial host [3]. Most recently, and perhaps now the most widely adopted commercial approach, CRISPR-Cas-mediated phage engineering is being developed [11].
Phico Therapeutics’ concept for its SASPject technology, namely rationally designed engineering of bacteriophages as delivery vehicles for use in the treatment of bacterial infections, came to fruition in 2000. SASPject utilises small acid soluble spore proteins (SASPs) as the antibacterial agent with the gene encoding SASP delivered to target bacteria by engineered phage [12] (Figure 1). Spore forming bacteria produce α/β type SASPs and γ type SASPs [13]. The primary sequence of γ type SASPs has not been as highly conserved in evolution as has those of α/β type SASPs and they have no known function other than generating amino acids by their degradation during spore germination [13]. α/β type SASPs (hereinafter globally referred to as SASP(s)) comprise a small family of proteins that provide, or contribute to, protection of spore DNA from environmental stresses such as UV irradiation, heat, desiccation, and general temporal decay [14,15,16]. SASPs bind to and saturate the whole spore genome forming a SASP-DNA complex whereby a myriad of H-bonding between SASP and phosphate, ribose, and nucleobase groups, along the minor grove of DNA results in the conformation of bacterial DNA shifting from the normal B-like towards A-like [17]. In spores, SASP binding is protective, but an inability to degrade SASP prevents spore outgrowth and vegetative cell survival [18,19]. This natural and sequence-independent binding by SASPs means they can likely be considered inter-changeable with pan spectrum activity. It is also unlikely that resistance will develop to SASP activity since mutations in bacterial DNA do not affect the ability of SASP to bind to and inactivate DNA. Phico has developed a SASPject engineered phage for the intranasal decolonisation of Staphylococcus aureus, including MRSA (SASPject PT1.2).
This article focuses on the pre-clinical microbiology programme conducted with PT1.2, which provides early exemplification of the potential validity and specificity of the SASPject technology for the treatment of bacterial infections. Phico has taken PT1.2 into the clinic in a Phase 1 trial demonstrating a good safety and tolerability profile [20].
2. Results
2.1. Design of PT1.2
PT1.2 was engineered using rational design techniques from the wild type temperate S. aureus phage, phi11. The phi11 phage was selected because the full genome sequence was available (Genbank AF424781.1), it had no apparent propensity to carry toxin genes, and it was tractable to manipulate. In 2000, when methods to manipulate and, more particularly, select, engineered lytic phage were limited, the use of a temperate phage was considered optimal. Engineering could be simply carried out with phi11 as a prophage and the modified phage could be maintained in a suitable S. aureus host so that it could be manufactured from a single master cell bank, rather than requiring an additional phage bank. S. aureus strain 8325-4 was selected as the final host strain for PT1.2 as it has been designated by the UK Health and Safety Executive as category 1, making it an ideal strain for manufacturing, particularly at large scale. Conventional cloning and allelic exchange techniques were employed to rationally design the engineered PT1.2, with positive selection for modified phi11 enabled by insertion of a gene encoding cadmium resistance downstream of the SASP gene and selection on CdCl2 agar plates. The SASP-C gene from Bacillus megaterium was used in conjunction with a constitutive S. aureus promoter, fbaA. This SASP expression cassette replaced the phi11 native holin gene and was inserted in the opposite orientation to the phi11 late genes. Deletion of the holin gene and thus eliminating the presence of holin within target bacteria means that the phage endolysins cannot access the bacterial cell wall and cause lysis. Inhibiting the ability of PT1.2 to lyse targeted S. aureus bacteria was considered desirable since it enabled production of SASP to continue within targeted cells and also inhibited the release of any toxins or toxin/antibiotic resistance genes into the infection environment. Similarly, prevention of lysis minimised the release of inflammatory S. aureus cell wall components that are caused by phage-mediated lysis. The extended production time of SASP was designed to maximise its coverage of the target S. aureus genome and plasmid DNA: preliminary data suggests SASP-bound plasmid DNA is not taken up by other cells, which may help to limit the spread of plasmid-borne antibiotic resistance genes (data not shown). The development of PT1.2 is shown in Figure 2.
2.2. Spectrum of Activity and Specificity of PT.2
All 225 isolates of S. aureus tested showed susceptibility to SASPject PT1.2 using a range of methods comprising kill curves, or no growth of targeted cells 3 h after exposure to PT1.2 in microtitre plate assays (Table 1). In the 3 h kill assay 153/163 (93.7%) MRSA and 23/34 (67.6%) MSSA isolates showed a 4 log10 or greater kill (a drop from 105 cfu/mL to 10 or fewer cfu/mL), with no viable cells observed at 3 h [21]. In time-kill curves, at the 4 h time point there were 15/24 (63%) MRSA and 2/4 (50%) MSSA isolates showing a 4 log10 or greater kill. Overall 177/187 (94.6%) MRSA and 29/38 (76.3%) MSSA isolates showed a ≥3 log10 kill by 3 or 4 h.
Among the 10 isolates of MRSA where a less than 3 log10 reduction in viable count was achieved, 4 strains (7079, 88032, 17047 from the UK and 7050 from the USA) had no further distinguishing details, whilst the other 6 strains (160006, 100096 from the UK, 160002 from Japan and strains 14545, 15818, 24764 of unknown geographical origin) exhibited intermediate resistance to vancomycin. All 12 Panton-Valentine Leukocidin
(PVL)-positive MRSA and MSSA strains exhibited a 4 log10 or greater kill within 3 h.
In the specificity studies none of 24 tested and confirmed coagulase-negative staphylococci showed any reduction in cell counts, confirming PT1.2 specificity (Table 2).
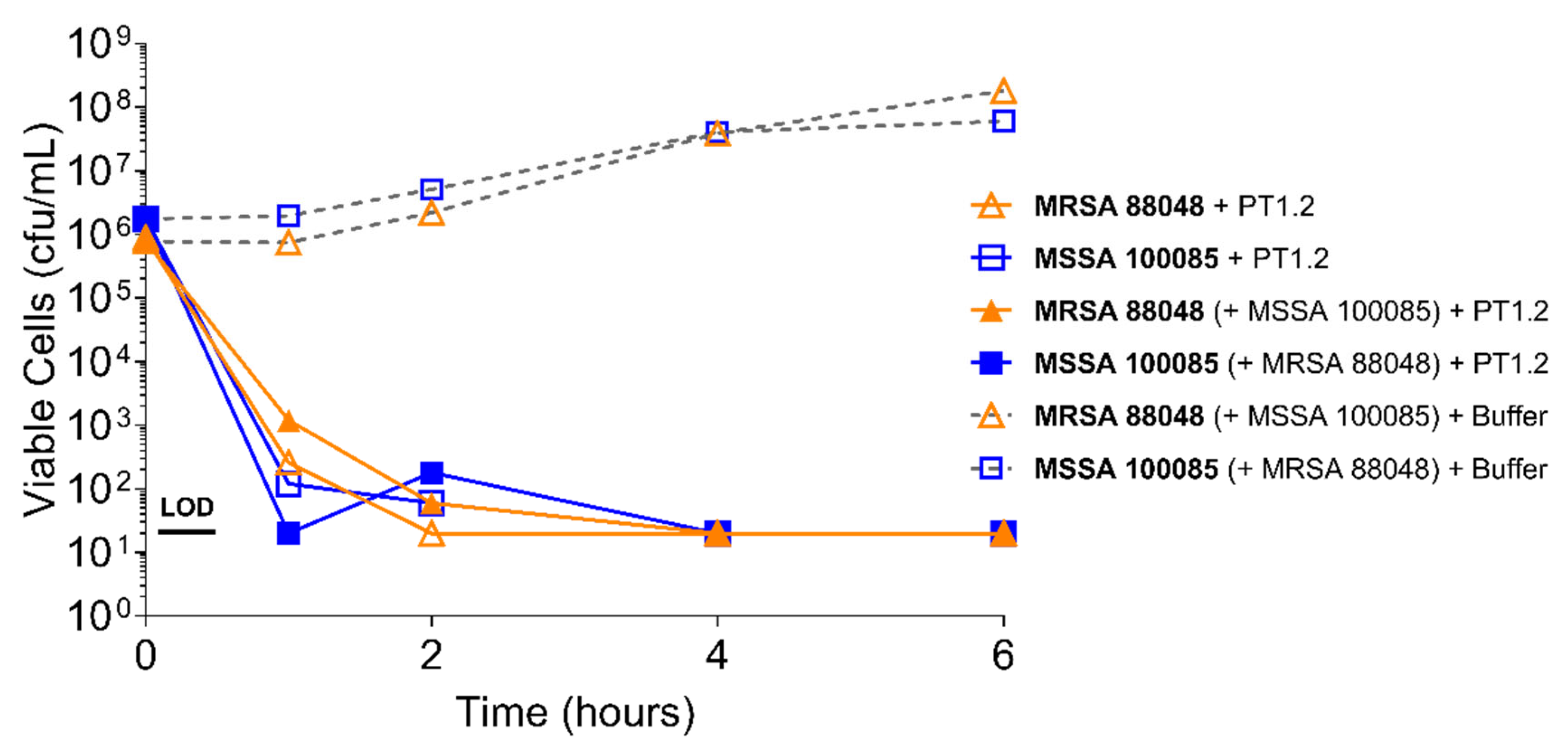
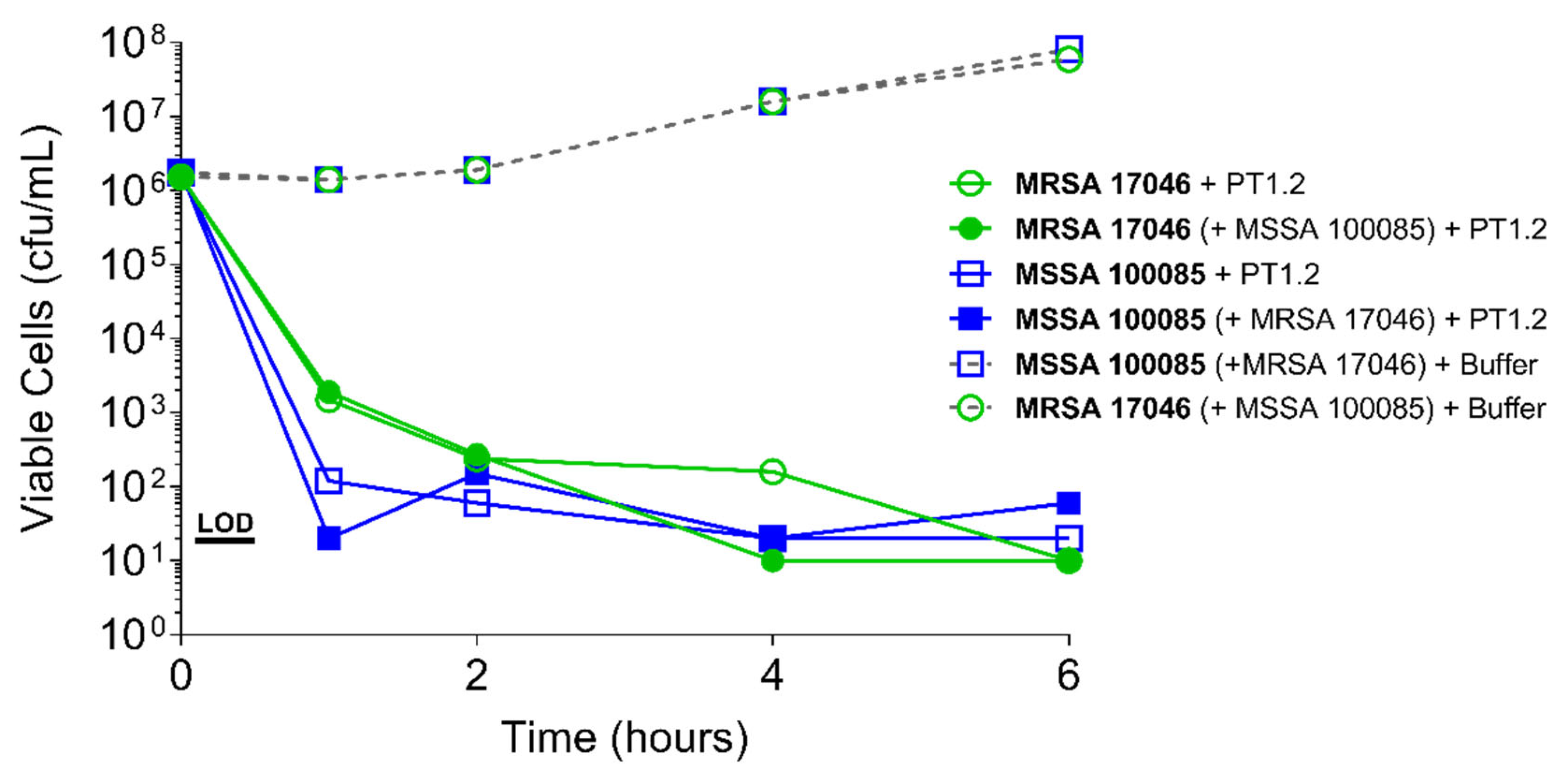
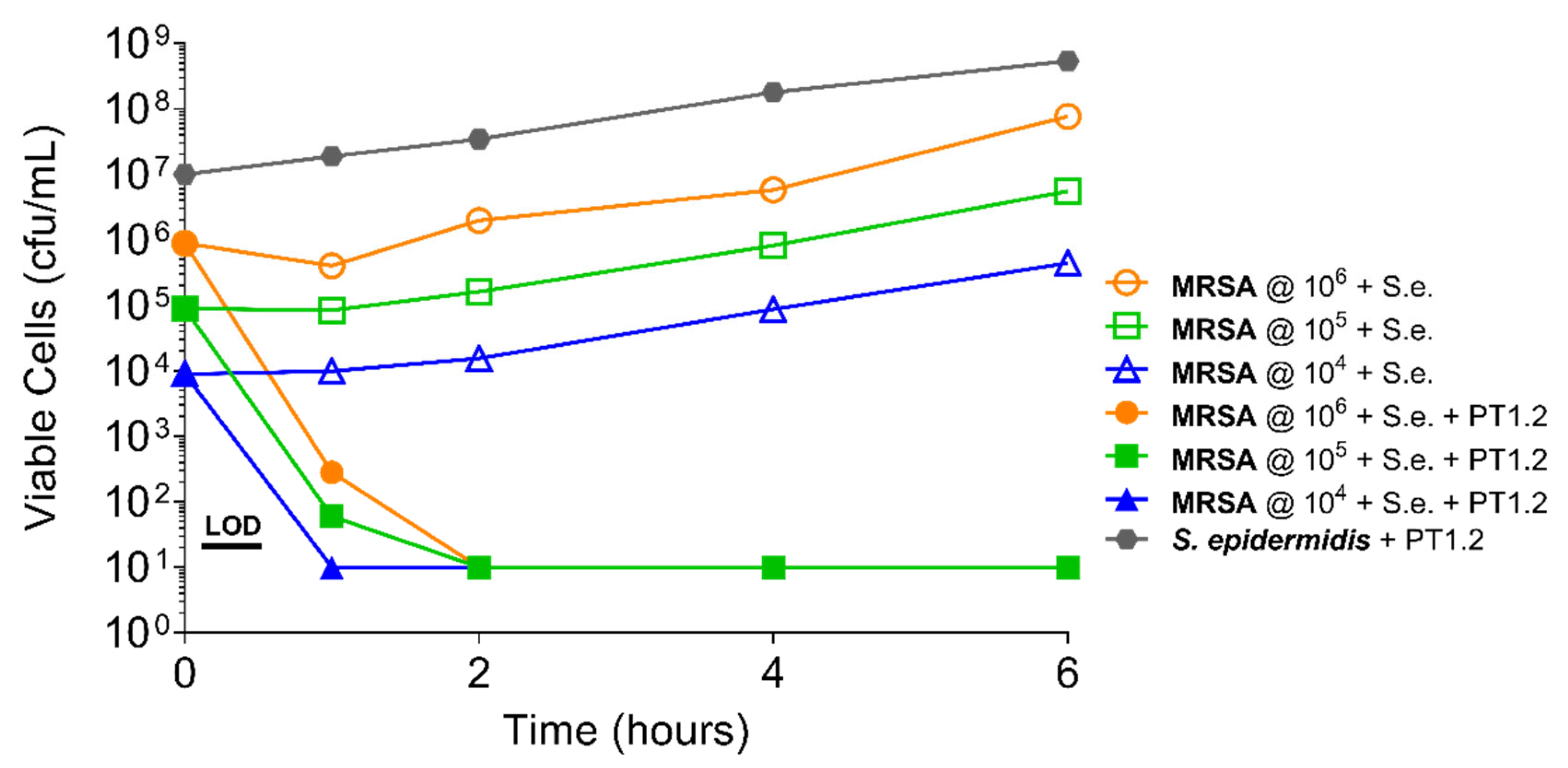
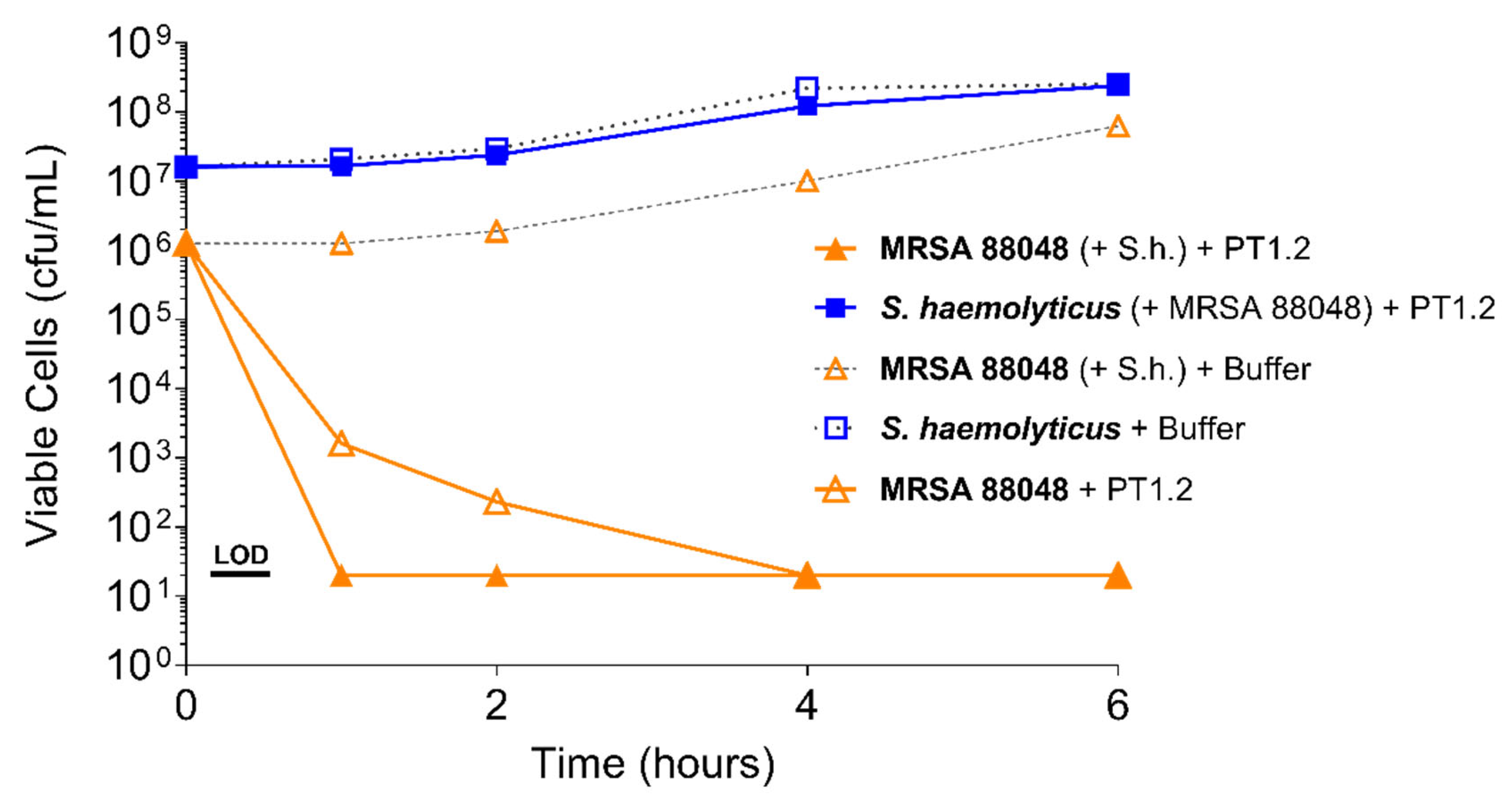
PT1.2 was equally effective against both MRSA strain 88048 and MSSA strain 100085 in mixed culture (Figure 3), as well as MRSA strain 17046 and MSSA strain 100085 in mixed culture (Figure 4), showing a 4 log10 or greater reduction in cell viability within 2 or 3 h, respectively. Activity against these strains in co-culture did not differ materially from the activity of PT1.2 against each strain alone although the rate of kill of the MSSA strain in co-culture was slightly faster than for either MRSA strain. Likewise the presence of S. epidermidis strain 72020 at 107 cfu/mL in co-culture with MRSA 88048 at cell concentrations ranging from 104–106 cfu/mL had no effect on activity of PT1.2 (Figure 5). A > 4 log10 drop in cell viability was seen for all MRSA 88048 cultures (both alone and in co-culture) within 2 h of addition of PT1.2. A co-culture of S. haemolyticus strain 133034 (107 cfu/mL) with MRSA strain 88048 (106 cfu/mL) also had no effect on PT1.2 activity against the MRSA (Figure 6).
2.3. Speed of Kill
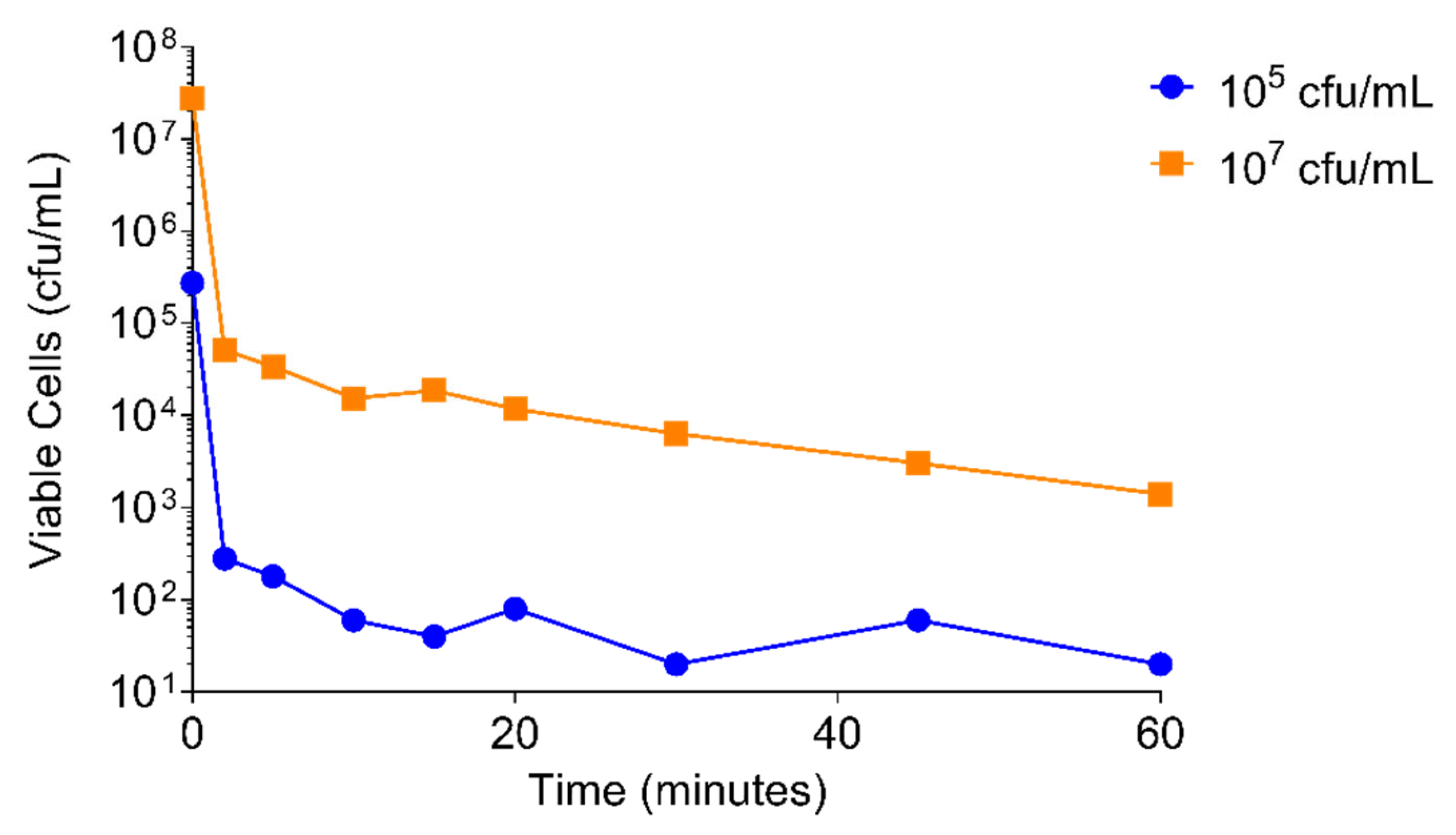
A 3 log10 drop in viability of S. aureus USA300 was achieved within 2 min of adding PT1.2 to a culture of 105 cfu/mL and within 10 min of addition to a culture of 107 cfu/mL (Figure 7).
2.4. Kinetics of PT1.2:S. aureus Interaction
2.4.1. PT1.2 Concentration
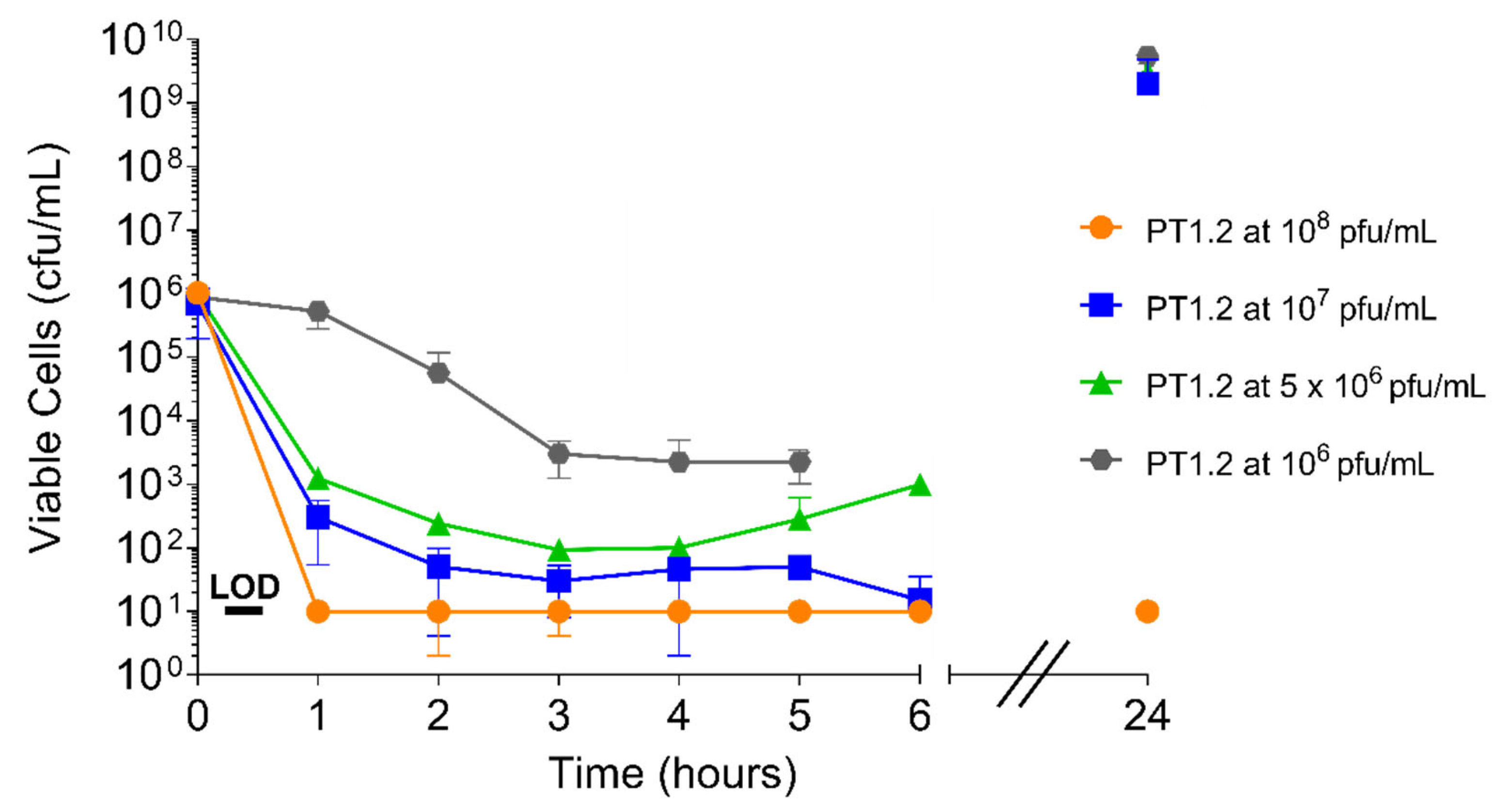
Both the speed and extent of activity of PT1.2 against S. aureus 02ST4127 were affected by PT1.2 concentration (Figure 8). A reduction in viable cell numbers to below the limit of detection (10 cfu/mL) within 2 h with PT1.2 at 108 pfu/mL was consistent for all triplicate cultures. However, there was marked variation in the speed and extent of kill between triplicate cultures with all other concentrations of PT1.2. For all concentrations except 108 pfu/mL, where viable cell numbers remained below the limit of detection, there was varied regrowth of surviving cells at 24 h.
2.4.2. S. aureus Concentration
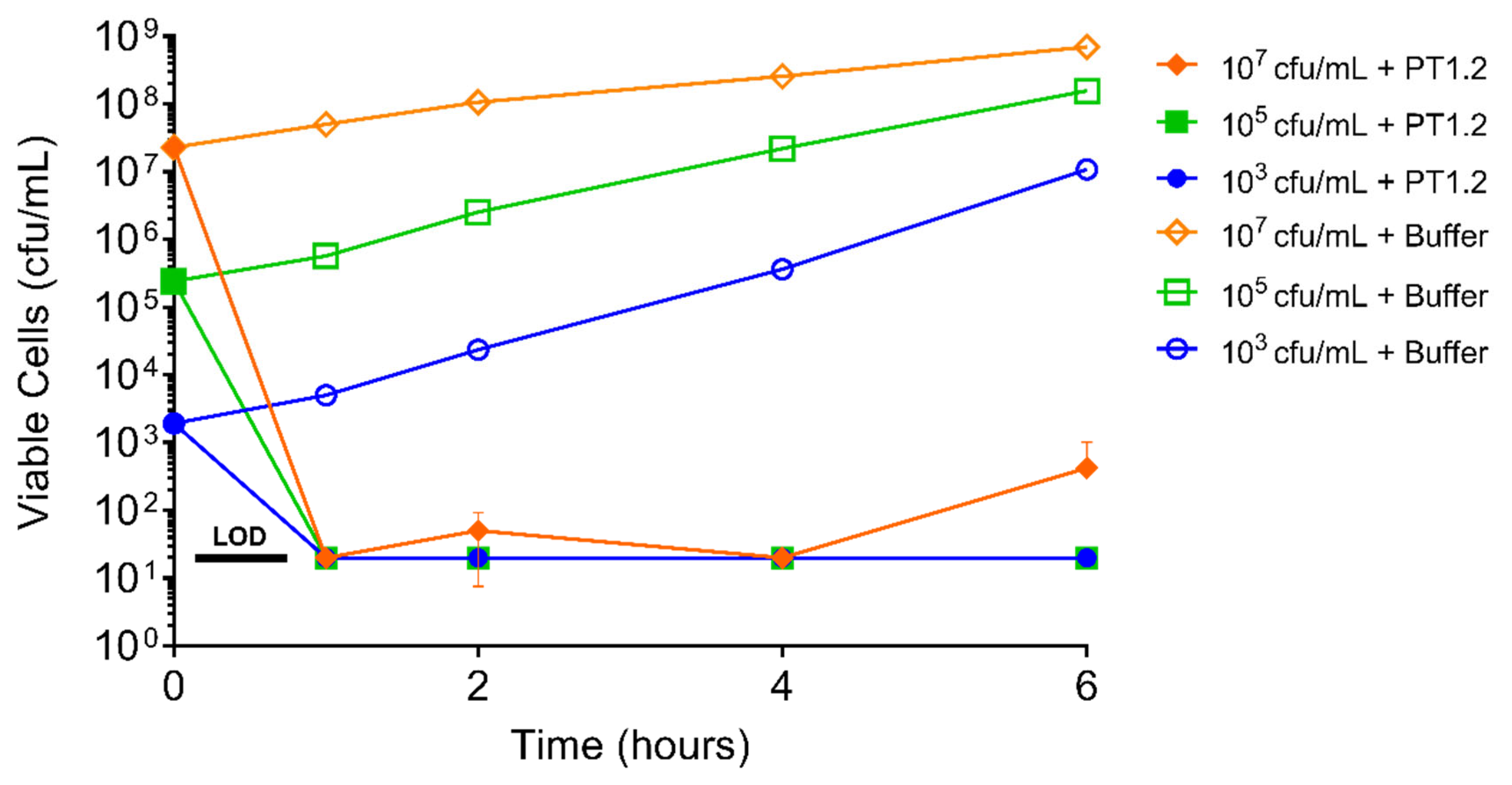
PT1.2 exhibited a rapid kill against S. aureus USA300 at concentrations spanning 1.5 × 103 to 1.5 × 107 cfu/mL showing a drop in viable cell numbers to below the limit of detection (20 cfu/mL) within 1 h for all cultures (Figure 9).
2.5. Effect of Human Serum Albumin on PT1.2 Activity
The activity of PT1.2 against both S. aureus USA300 and EMRSA-16 was not compromised by the presence of HSA (50 g/L) (Figure 10). A 5-log10 or greater reduction in viable cell numbers was observed within 1 h in the presence and absence of HSA.
2.6. Assessment of S. aureus Resistance to PT1.2
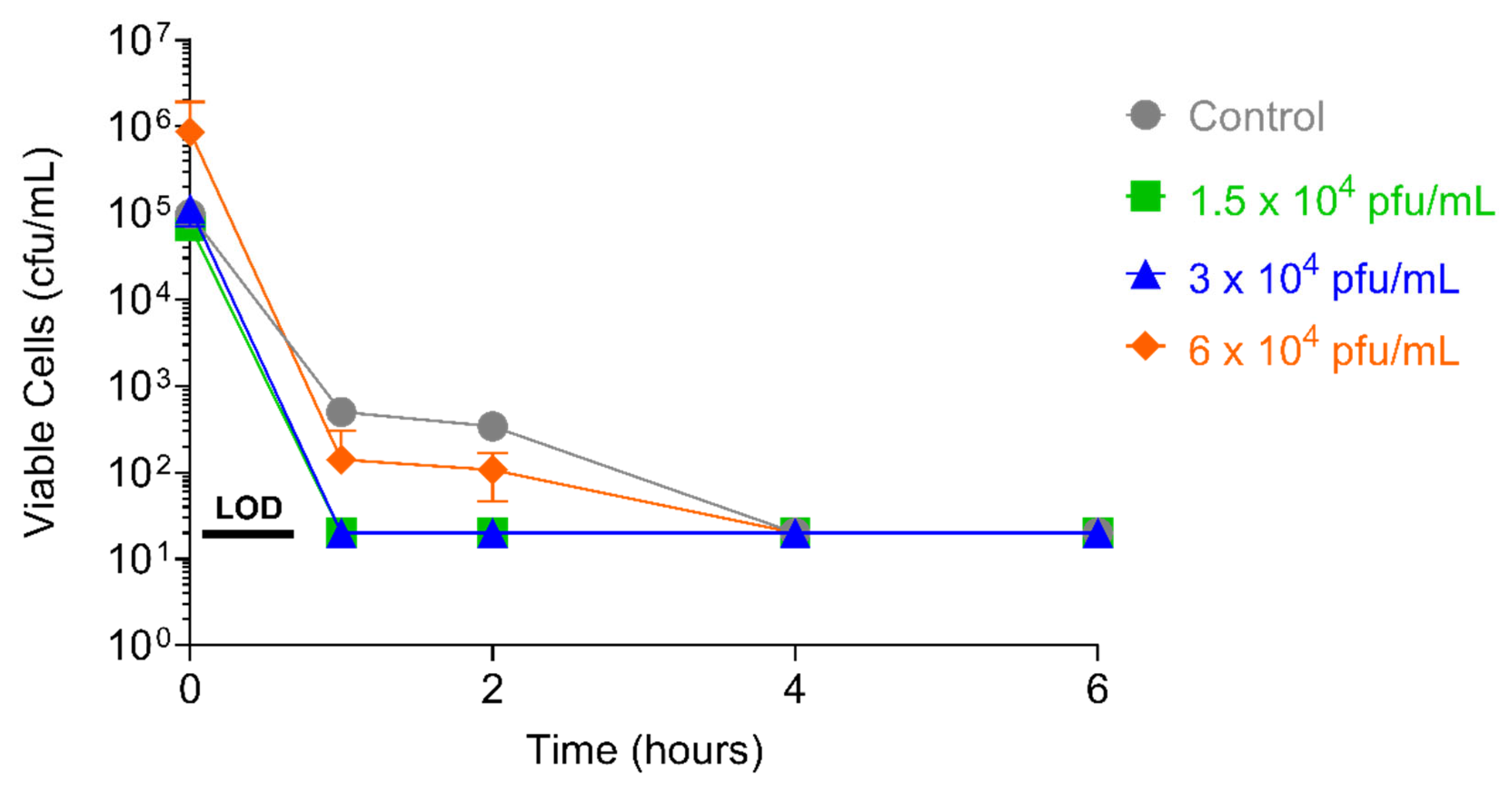
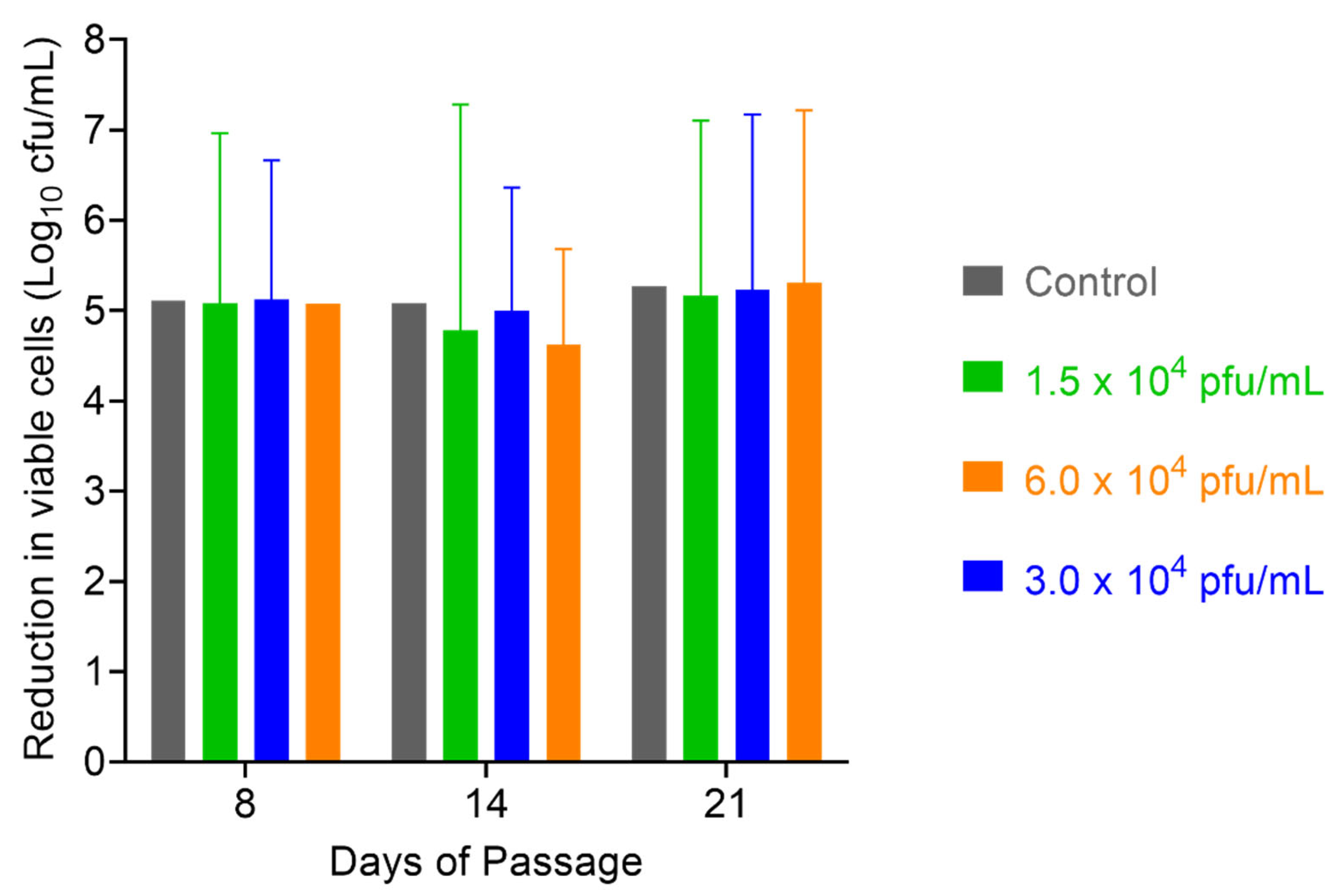
After 52 days of serial passaging, when the study was terminated (46 days for the 1 × 104 pfu/mL dose of PT1.2), the susceptibility of EMRSA strain O2ST4285 to PT1.2 remained equal to that observed for the control (Figure 11). For S. aureus USA300 no consistent or significant shift in susceptibility to PT1.2 was seen during passaging over the 21 days of the experiment (Figure 12). In the single dosing study, no PT1.2-resistant isolates of S. aureus USA300 were found amongst the 1.2 × 108 cfu tested.
3. Discussion
Phage therapy has been undergoing a resurgence in recent years driven, at least in part, by the “next generation phage therapy” that comprises engineered phage. Engineering can facilitate precision control of important considerations such as host range, manufacturability, and PK profile, for example, by transferring the genetics of these traits at optimum into a single or small group of phages making up a drug product. Engineering enables a move away from the large cocktails of diverse phages traditionally used in wild type phage therapy by concentrating these attributes across much fewer, ideally related, phages.
Phico engineers phages to deliver a gene encoding a small acid soluble spore protein, SASP, to target bacterial species. The SASP gene is placed under the control of a constitutive promoter from the target bacterium so that, following injection of the SASP gene, intracellular expression is immediate and continual. SASPs bind in a non-sequence specific manner via two highly conserved regions (AIDQMKYEIASEFGVNLG and TSRANGSVGGEITKRLVQM) resulting in a shift in DNA conformation from the normal B-like to A-like [16,17]. The amino acid sequences of SASPs show exceptional conservation both within and across species of endospore-forming bacteria, both aerobic and anaerobic, yet they show minimal homology to bacterial gene sequences in current databases [16]. Uniquely, in view of their mode of action, SASP also bear no sequence similarity to other DNA-binding proteins in available databases and contain no known sequence motifs, including motifs found in other DNA-binding proteins [16]. Expressed in vegetative bacteria, SASPs act to inactivate the bacterial DNA by preventing DNA replication and, where bound, transcription.
SASPject PT1.2 comprises a single temperate phage in which the holin gene has been removed and replaced with a SASP gene expression cassette containing the SASP-C gene from Bacillus megaterium under the control of constitutive S. aureus promoter, fbaA. PT1.2 is derived from S. aureus phage, ϕ11. PT1.2 has been developed for the intranasal decolonisation of S. aureus, including MRSA and is in further pre-clinical development for systemic applications. Given that 33% of surgical site infections (SSIs) can be caused by S. aureus [22], evidence that nasal S. aureus decolonisation can lead to a decreased incidence of S. aureus infection has led to this practice being implemented on a wide scale, and recommended, in conjunction with chlorhexidine body washing, by the UK National Institute for Health and Care Excellence [23]. There remain limited options for effective nasal decolonisation of S. aureus; mupirocin is the primary agent of choice although resistance rates of up to 9% have been reported [24]. The use of other antibiotics is also potentially impacting on mupirocin efficacy with a study showing that sub-MIC levels of ciprofloxacin increase adhesion of quinolone-resistant MRSA, perhaps explaining persistent MRSA colonization and failure of mupirocin treatment in patients who received a fluoroquinolone [25,26]. That mupirocin remains the most effective antibiotic for MSSA and MRSA decolonisation, both in patients and healthcare personnel, means a reduction of its effectiveness presents a risk for invasive infection [24]. Hence, there remains a need for a new topical agent with potent efficacy, ideally bactericidal, and a low propensity for resistance development.
SASPject PT1.2, targeted at S. aureus, including MRSA has undergone a programme of pre-clinical microbiology testing that supported testing of PT1.2 in a Phase 1 clinical trial. PT1.2 shows rapid bactericidal activity against a wide range of geographically and genetically diverse S. aureus, including strains that are methicillin resistant and susceptible, vancomycin intermediate and heterologous vancomycin intermediate resistant, and both Panton-Valentine Leukocidin positive and negative [27,28,29,30]. Indeed, almost 95% (177/187) of MRSA and just over 76% (29/38) of MSSA isolates exhibited at least a 3 log10 kill within 4 h of addition of PT1.2. Remaining isolates were susceptible to SASPject PT1.2, but the killing rates were less impressive. No activity was evident against 24 coagulase-negative staphylococci representing nine species, confirming a high degree of target pathogen specificity. Many antibacterial agents, including existing “narrow spectrum” conventional agents will eliminate a significant proportion of the patient’s normal microbiota, leaving the body more susceptible to colonisation or infection by pathogens such Neisseria meningitidis or Streptococcus pneumoniae, for example, in the nose. As SASPject technology can be specifically targeted at selected pathogens, its use should have a minimal effect on the body’s normal bacterial microbiota and may result in fewer secondary infections or the selection of resistant colonising bacteria.
A key feature of the antibacterial component of SASPject, SASP, is the rapidity of its activity against S. aureus. Typically, for conventional antibiotics, rate of kill studies involve sampling at hourly intervals seeking to establish a 3 log10 reduction in viable cell count over 24 h [31]. SASP can achieve up to a 3 log10 drop in cell viability within 10 min in a 107 cfu/mL culture, and a 6 log10 reduction within 1 h against actively growing cells [24]. SASP is also rapidly bactericidal against stationary phase S. aureus (data not shown).
As the delivery of the SASP gene by SASPject delivery vectors is a truly novel approach to antibacterial therapy, it is expected to be unaffected by existing antibiotic resistance mechanisms. Indeed, in addition to the data given above for methicillin and intermediate vancomycin resistance, SASPject PT1.2 is minimally affected by all tested resistance mechanisms: penicillin, tetracycline, ciprofloxacin, rifampicin, erythromycin, fusidic acid, and gentamycin [23]. The rate of kill against some strains, including some, but not all, VISA strains, can show more variation and be generally slower. However, the mechanism for this is not clear and warrants further investigation.
That PT1.2 activity is unaffected by the presence of human serum albumin is encouraging and suggestive that protein:protein interactions in the body will not impact efficacy; the absence of any measurable binding to HSA is also positive for future systemic applications.
Due to the unique mode of action of SASP-binding to bacterial DNA irrespective of sequence, the likelihood of resistance developing to SASP is judged to be very low. Germinating spores clearly need to release their genome from SASP in order to outgrow. SASP degradation during spore germination is initiated by a single protease, GPR [32,33,34]. This enzyme is an endoprotease specific for a pentapeptide sequence found once or twice in all SASP. GPR is initially synthesized during sporulation and is processed twice to create a 40-kDa form, which then acts to initiate SASP degradation in vivo. Crucially, the activity of GPR is dependent on the conditions inside a germinating spore, i.e., low water activity, low pH, and the presence of dipicolinic acid, collectively, conditions that are not possible in vegetative bacteria [35]. However, the possibility of resistance arising to the SASPject phage to prevent injection of the SASP gene into target bacteria must also to be considered. Resistance to wild type viral vectors is a widely acknowledged phenomenon. However, the presence of a constitutively expressed SASP, which remains active independently of the fate of the phage delivery vehicle inside target cells, renders ineffective all of the intracellular resistance mechanisms that bacteria use against wild type viral vectors. A remaining resistance mechanism occurs at the stage of phage vector binding to the bacterial cell surface.
It has been suggested that staphylococcal phages bind to a common, and historically highly conserved, cell wall receptor [36,37]. This suggestion has been supported by the studies reported here, showing PT1.2 activity against all tested S. aureus strains. Furthermore, serial passaging studies of between 46 and 52 days, with 9 cultures of S. aureus strain O2ST4285 (EMRSA-16) and PT1.2 at inhibitory and sub-inhibitory levels, showed no consistent trend towards resistance to phage binding [38]. No naturally occurring resistance was observed in the >1 × 108 tested S. aureus cells. That the limiting factor deciding host spectrum of S. aureus phages occurs post-injection of phage vector DNA has been borne out by studies, confirming that SASPject PT1.2 is effective against isolates (including EMRSA-15 types), which do not support the parental wild type viral vector (data not shown).
A theme in some of the kill curve studies reported here is that of occasional regrowth of target cells to highly variable levels. Since remaining viable cells do not exhibit resistance when used in further time-kill assays, one or more other factors are influencing the outcome. Compared to the number of molecules of a conventional antibiotic, even at sub-MIC level, very few phage particles are used. As such, limited physical interaction between phage vector and bacteria is likely to be a contributory factor and this phenomenon is currently being investigated further.
Overall, important considerations for a commercial antibacterial agent using bacteriophage-based technologies include host range, manufacturability, pharmacokinetics, and stability. All, with perhaps the exception of stability, have the potential to be improved by engineering of phages. Host range can be broadened by engineering phages that express a multiplicity of host binding domains to steer away from extensive phage cocktails and limit the number of phages that make up a product, or engineering different versions of a single lead phage each expressing a modified or alternative host binding domain [39,40,41,42]. Limiting the number of phages in a product simplifies the issue of pharmacokinetics where different phages can have widely varying half lives in the body making judgements about dosing intervals/regimens difficult; again, this has potential to be controlled/improved by phage engineering [43]. For manufacturing, the fewer phages per product, the better, as each phage can have its own production and purification requirements, making the process to a drug product more challenging to control and show robustness and reproducibility. In addition, the final step of mixing the phages to make the product, in exact, consistent ratios whilst maintaining sterility, introduces greater risk as the number of different phages increases. Missteps at this stage can lead to wastage of batches driving up cost of goods. Thus, phage engineering, with the promise of limiting phage numbers, can have a major impact on simplifying manufacturing. Stability is a key consideration, and the importance of this was demonstrated in a phage therapy clinical trial for the topical treatment of P. aeruginosa infected burns [44]. This so-called Phagoburn study involved treatment with cocktails of up to twelve P. aeruginosa phages at a planned dose of 106 pfu/mL. The study was halted prematurely due to failure to show activity, and it was subsequently found that the active titre of the phage preparation had dropped to 101–102 pfu/mL.
Commercially, phage therapy remains in its infancy with infrequent compassionate-use cases [45,46,47]. To become a more widespread treatment option, well-designed clinical trials, using phages that are the product of robust, reproducible, well-controlled manufacturing processes, need to be executed with both engineered and wild type phages.
4. Materials and Methods
4.1. The Strains, Media and Chemicals
MRSA USA300 strain 43484 and EMRSA-16 strain 252 were routinely used for time-kill curves, except where stated otherwise.
Overnight cultures were grown in Luria-Bertani (LB) broth (5 mL) in sterile Universal tubes, incubated at 37 °C with shaking (350 rpm) and diluted for time-kill curves in LB broth containing CaCl2 (10 mM), pH 7.2 (LBC broth). SASPject PT1.2 suspended in Tris buffered saline (50 mM) containing CaCl2 (10 mM) and MgCl2 (1 mM) (ΦTBS) was used at a standard, final concentration of 1.5 × 108 pfu/mL, unless stated otherwise.
Controls were performed for each experiment where PT1.2 was substituted with an equivalent volume of ΦTBS.
4.2. Spectrum of Activity of PT1.2
4.2.1. 3 h Kill Assay
See Table 1 for characteristics of S. aureus isolates. Cultures were grown to mid-8 log10 phase and diluted to ~2 × 105 cfu/mL in LBC broth. Diluted culture (55 μL) was added to PT1.2 (55 μL) in a microtitre well. Growth controls for each strain were set up where PT1.2 was replaced with ΦTBS. Aliquots of 100 μL were removed from each sample well after 3 h incubation at 35 °C and spread onto blood agar plates. Colony counts were carried out after overnight incubation at 35 °C.
4.2.2. Time-Kill Curves
Cultures (see Table 1 for isolate characteristics) were grown overnight and diluted to a final inoculum of 105 cfu/mL with reference to a McFarland standard. PT1.2 was added to the diluted cultures. Tubes were incubated at 35–37 °C without shaking and aliquots (100 μL) were removed at 0, 1, 2, 4, 6, and 24 h and spiral plated (50 μL) onto nutrient agar plates or blood agar plates. Colony counts were carried out after overnight incubation at 35–37 °C.
4.3. Specificity of PT1.2
4.3.1. Activity of PT1.2 against Coagulase Negative Staphylococci
The 3 h kill assay was also conducted with twenty four isolates (Health Protection Agency (HPA) Cambridge identity codes (CC) given) of the staphylococcal species listed in Table 3.
4.3.2. Activity of PT1.2 in Mixed Bacterial Cultures
Isolates used for mixed culture kill assays are listed in Table 4.
MSSA/MRSA co-cultures: MSSA/MRSA cultures were diluted to a final inoculum of 4 × 106 cfu/mL. In Universal tubes, 200 μL of MSSA culture was mixed with an equal volume of MRSA culture and 400 μL of PT1.2 was added, yielding final concentrations of 106 cfu/mL for each bacterial strain and 1.5 × 108 pfu/mL of PT1.2. In addition to a ϕTBS control, LBC broth (200 μL) was added to a mono-culture of each strain (200 μL) plus PT1.2 (200 μL).
MRSE/MRSA co-culture: MRSE was diluted to 4 × 107 cfu/mL from an overnight culture, and MRSA strain 88,048 was diluted to 4 × 106, 4 × 105 and 4 × 104 cfu/mL from an overnight culture. In Universal tubes, 200 μL of diluted MRSE culture was mixed with an equal volume of MRSA culture and 400 μL PT1.2 was added. This yielded final concentrations of 107 cfu/mL MRSE with either 106, 105, or 104 cfu/mL MRSA and 1.5 × 108 pfu/mL of PT1.2. In addition to a ϕTBS control, MRSE was replaced by an equal volume of LBC broth (200 μL) to give MRSA only.
MRSH/MRSA co-cultures: Overnight cultures of MRSH and MRSA strain CC 88,048 were diluted to 4 × 107 cfu/mL and 4 × 106 cfu/mL, respectively. In Universal tubes, 200 μL of diluted MRSH culture was mixed with an equal volume of MRSA culture and 400 μL PT1.2 was added. This yielded final concentrations of 107 cfu/mL MRSH, 106 cfu/mL MRSA and 1.5 × 108 pfu/mL of PT1.2. In addition to a ϕTBS control, MRSH was replaced by an equal volume of LBC broth (200 μL) to give MRSA only.
All samples were incubated at 35 °C without shaking and aliquots (150 μL) were removed at 0, 1, 2, 4, and 6 h and spiral plated (50 μL) onto both Colombia blood agar plates and chromID MRSA plates (Biomerieux). Colony counts were carried out after overnight incubation at 35 °C with total cell numbers enumerated on Colombia blood agar plates and MRSA cells enumerated on chromID plates. The number of non-MRSA surviving colonies was calculated by subtracting the cfu/mL derived from each chromID MRSA plate from the number derived from the corresponding Colombia blood agar plate.
4.4. Effect of PT1.2 on Bacterial Viability
4.4.1. Speed of Kill
S. aureus MRSA USA300 was grown overnight at 37 °C without shaking, and then diluted to a final concentration of either 2.7 × 107 or 2.7 × 105 cfu/mL. PT1.2 was added to each culture dilution. Cultures were incubated at room temperature without shaking and aliquots (100 μL) were removed at 0, 2, 5, 10, 15, 20, 30, 45 and 60 min and spiral plated (50 μL) onto blood agar plates. Colony counts were enumerated after overnight incubation at 37 °C.
4.4.2. Concentration of PT1.2
An overnight culture of EMRSA-15 strain 02ST4127 was diluted to 4 × 106 cfu/mL and aliquots mixed with an equal volume of PT1.2 to give final concentrations of 108, 107, 5 × 106 and 106 pfu/mL. Cultures were incubated at 37 °C without shaking and aliquots (100 μL) were removed at 0, 1, 2, 3, 4, 5, 6, and 24 h and plated (100 μL) onto LB agar plates. Colony counts were enumerated after overnight incubation at 37 °C.
4.4.3. Concentration of Bacterial Culture
Time-kill curves were carried out with PT1.2 added to overnight cultures of S. aureus EMRSA-15 strain O2ST4127 each diluted giving final concentrations of 1.5 × 107, 1.5 × 105 and 1.5 × 103 cfu/mL. Cultures were incubated at 37 °C without shaking and aliquots (100 μL) were removed at 0, 1, 2, 4 and 6 h and spiral plated (50 μL) onto LB agar plates. Colony counts were enumerated after overnight incubation at 37 °C.
4.5. Effect of Human Serum Albumin (HSA) on PT1.2 Activity
S. aureus USA300 and EMRSA-16 were grown overnight and then each diluted to 2 × 107 cfu/mL. PT1.2 was added to 2 × 107 pfu/mL (final concentration) and after removing a T0 (100 μL) aliquot, HSA was added to a final concentration of 50 g/L. The samples plus controls were incubated without shaking at 37 °C, and aliquots (60 μL) removed at 1, 2, 4, and 6 h. All aliquots were spiral plated (50 μL) on LB agar plates. Colony counts were performed after overnight incubation at 37 °C.
4.6. Assessment of S. aureus Resistance to SASPject PT1.2
4.6.1. Passaging Studies
An EMRSA-16 strain O2ST4285 culture was grown overnight at 37 °C. The culture was diluted to 105 cfu/mL (final concentration) and PT1.2 was added to final, sub-inhibitory concentrations of 1.5 × 104, 3 × 104 or 6 × 104 pfu/mL, each in triplicate. Triplicate controls, with an equal volume of LBC broth replacing PT1.2, were also set up. Cultures were grown overnight at 37 °C without shaking. Daily for 52 successive days (46 days for cultures with 1.5 × 104 pfu/mL PT1.2) aliquots of sample and control cultures were diluted to 105 cfu/mL, PT1.2 was added as described above, and again grown overnight. The susceptibility of the cultures to PT1.2 was assessed by time-kill assay at weekly intervals: 105 cfu/mL of each passaged culture and 1.5 × 108 pfu/mL PT1.2 were mixed and incubated at 37 °C without shaking. Aliquots (75 µL) were removed at 1, 2, 4, and 6 h and spiral plated (50 µL) onto LB agar plates and colonies enumerated after overnight incubation at 37 °C. This study was repeated with S. aureus USA300 for a period of 21 days except susceptibility to PT1.2 was assessed on days 7, 14, and 21 at a single 4 h time point.
4.6.2. Single Dosing Study
In order to estimate the frequency of any naturally occurring resistance to PT1.2, a culture of S. aureus USA300 (1.2 × 107 cfu/mL) was aliquoted into 10 × 1 mL fractions and centrifuged. Each pellet was resuspended in LBC broth containing PT1.2 (1.5 × 108 pfu) and incubated for 4 h at 37 °C, and then spread on to LB agar plates and incubated overnight at 37 °C. Any colonies which grew were re-tested for susceptibility to PT1.2 by time-kill curve as described above.
5. Conclusions
In summary, SASPject PT1.2 has provided promising in vitro data against S. aureus to support its further development as well as demonstrating the potential of SASPject as a novel antibacterial platform technology. Given the expected pan spectrum antibacterial activity of SASP, coupled with the ubiquity of phages for a target bacterial species, SASPject technology has the potential to be directed against any selected Gram positive or Gram negative bacterial target. SASPject PT1.2 demonstrates rapid bactericidal activity and is effective against all of the diverse range of S. aureus isolates tested, whilst showing complete specificity with activity unaffected by the presence of other staphylococcal species. SASPject PT1.2 showed a low propensity for resistance to develop. SASPject PT1.2 and the SASPject platform has the potential to provide a new therapeutic approach and a novel class of biological antibacterial that can be selectively targeted to individual or multiple bacterial species or genera.
Author Contributions
A.B. contributed to the experimental work and to review and editing. J.C. contributed to review and editing. H.F. prepared the original draft, supervised the work, and acquired the funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by private investment.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
Phico Therapeutics would like to acknowledge the following people for their contribution to the experimental work described in this article: Former employees, Adam Wilkinson, Katy Pitts, Vidya Math, Sarah Holme, Hongmei Wang, Alison Jackson, and Iona Kendall. Karen E. Bowker and Alasdair P. MacGowan of the Bristol Centre for Antimicrobial Resistance and Evaluation, Bristol, UK. Derek Brown of the Health Protection Agency at Addenbrooke’s Hospital, Cambridge UK.
Conflicts of Interest
James Cass is R&D Director for Phico Therapeutics. Anne Barnard is Research Manager of Phico Therapeutics. Heather Fairhead is CEO of Phico Therapeutics.
References
- Dheman, N.; Mahoney, N.; Cox, E.M.; Farley, J.F.; Amini, T.; Lanthier, M.L. An Analysis of Antibacterial Drug Development Trends in the United States, 1980–2019. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cully, M. Antibiotics alter the gut microbiome and host health. Nat. Res. 2019, 15, S19. [Google Scholar]
- Łobocka, M.; Dąbrowska, K.; Górski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 2021, 35, 225–280. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [Green Version]
- Brüssow, H.; Kutter, E. Phage Ecology. In Bacteriophages: Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, USA, 2005; Volume 70, pp. 129–163. [Google Scholar]
- Chanishvili, N. Phage Therapy—History from Twort and d’Herelle through Soviet experience to current approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [PubMed]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.R.; Oliveira, H.; Pires, D.P.; Dąbrowska, K.; Azeredo, J. Phage therapy efficacy: A review of the last 10 years of preclinical studies. Crit. Rev. Microbiol. 2020, 46, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Swigoňová, Z.; Balachandran, A.; Oldfield, L.M.; van Kessel, J.C.; Hatfull, G.F. BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef] [Green Version]
- Fairhead, H.; Wilkinson, A.; Barnard, A.; Severi, E.; Anderson, N.; Pitts, K. Modifying Bacteriophage. European Patent No. EP3201323, 6 November 2019. [Google Scholar]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairhead, H. SASP gene delivery: A novel antibacterial approach. Drug News Perspect. 2009, 22, 197–203. [Google Scholar] [CrossRef]
- Sanchez-Salas, J.-L.; Santiago-Lara, M.L.; Setlow, B.; Sussman, M.D.; Setlow, P. Properties of Bacillus megaterium and Bacillus subtilis Mutants Which Lack the Protease That Degrades Small, Acid-Soluble Proteins during Spore Germination. J. Bacteriol. 1992, 174, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Fairhead, H.; Setlow, B.; Setlow, P. Prevention of DNA damage in spores and in vitro by small, acid-soluble proteins from Bacillus species. J. Bacteriol. 1993, 175, 1367–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairhead, H.; Setlow, P. Binding of DNA to ac/d-Type Small, Acid-Soluble Proteins from Spores of Bacillus or Clostnidium Species Prevents Formation of Cytosine Dimers, Cytosine-Thymine Dimers, and Bipyrimidine Photoadducts after UV Irradiation. J. Bacteriol. 1992, 174, 2874–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setlow, P. I will survive: DNA protection in bacterial spores. Trends Microbiol. 2007, 15, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Bumbaca, D.; Kosman, J.; Setlow, P.; Jedrzejas, M.J. Structure of a protein–DNA complex essential for DNA protection in spores of Bacillus species. Proc. Natl. Acad. Sci. USA 2008, 105, 2806–2811. [Google Scholar] [CrossRef] [Green Version]
- Setlow, B.; Hand, A.R.; Setlow, P. Synthesis of a Bacillus subtilis small, acid-soluble spore protein in Escherichia coli causes cell DNA to assume some characteristics of spore DNA. J. Bacteriol. 1991, 173, 1642–1653. [Google Scholar] [CrossRef] [Green Version]
- Hayes, C.S.; Setlow, P. An alpha/beta-type small, acid-soluble spore protein which has a very high affinity for DNA prevents outgrowth of Bacillus subtilis spores. J. Bacteriol. 2001, 183, 3982–3990. [Google Scholar] [CrossRef] [Green Version]
- Hatzixanthis, K.; Wilkinson, A.; Fairhead, H. Double-Blind, Placebo-Controlled Phase I study of PT1.2, a Novel Antibacterial Protein (SASP) delivery vector. In Proceedings of the Interscience Conference on Antimicrobial Agents and Chemotherapy, Boston, MA, USA, 12–15 September 2010. Poster F1-2086b. [Google Scholar]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenks, P.J.; Laurent, M.; McQuarry, S.; Watkins, R. Clinical and economic burden of surgical site infection (SSI) and predicted financial consequences of elimination of SSI from an English hospital. J. Hosp. Infect. 2014, 86, 24–33. [Google Scholar] [CrossRef]
- NICE Guideline Updates Team (UK). Evidence Review for Effectiveness of Nasal Decolonisation in Prevention of Surgical Site Infection: Surgical Site Infections: Prevention and Treatment; NICE guideline NG125; National Institute for Health and Care Excellence: London, UK, 2019. [Google Scholar]
- Dadashi, M.; Hajikhani, B.; Darban-Sarokhalil, D.; van Belkum, A.; Goudarzi, M. Mupirocin resistance in Staphylococcus aureus: A systematic review and meta-analysis. J. Glob. Antimicrob. Resist. 2020, 20, 238–247. [Google Scholar] [CrossRef]
- Bisognano, C.; Vaudaux, P.; Rohner, P.; Lew, D.P.; Hooper, D.C. Induction of fibronectin-binding proteins and increased adhesion of quinolone-resistant Staphylococcus aureus by subinhibitory levels of ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1428–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbarth, S.; Liassine, N.; Dharan, S.; Herrault, P.; Auckenthaler, R.; Pittet, D. Risk Factors for Persistent Carriage of Methicillin-Resistant Staphylococcus aureus. Clin. Infect. Dis. 2000, 31, 1380–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, K.; Brown, D.F.; Barnard, A.; Wilkinson, A.; Fairhead, H. SASP: Rapid Bactericidal Activity against MRSA and Stationary Phase Staphylococcus aureus. In Proceedings of the Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Poster F1-2131. [Google Scholar]
- Bowker, K.E.; Noel, A.R.; MacGowan, A.P.; Pitts, K.; Wilkinson, A.; Fairhead, H. SASP: Kill Kinetics against Diverse Antibiotic Resistant Staphylococcus aureus. Proceeding of the Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007. Poster F1-2131. [Google Scholar]
- Barnard, A.; Pitts, K.; Brown, D.F.; Wilkinson, A.; Fairhead, H. SASP: Rapid Bactericidal Activity against USA strains of Methicillin Resistant Staphylococcus aureus (MRSA). In Proceedings of the European Congress on Clinical Microbiology and Infectious Diseases, Barcelona, Spain, 19–22 April 2008. Poster P-561. [Google Scholar]
- Pitts, K.; Barnard, A.; Brown, D.F.J.; Wilkinson, A.; Fairhead, H. The Efficacy of SASP Targeted to Methicillin Resistant Staphylococcus aureus (MRSA) in Mixed Staphylococcal Cultures. In Proceedings of the European Congress on Clinical Microbiology and Infectious Diseases, Barcelona, Spain, 19–22 April 2008. Poster P-561. [Google Scholar]
- National Committee for Clinical Laboratory Standards; Barry, A.L. Methods for Determining Bactericidal Activity of Antimicrobial Agents: Approved Guideline; M26-A. CLSI; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2009; Volume 19. [Google Scholar]
- Loshon, C.A.; Setlow, P. Bacillus megaterium spore protease: Purification, radioimmunoassay, and analysis of antigen level and localization during growth, sporulation and spore germination. J. Bacteriol. 1982, 150, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loshon, C.A.; Setlow, B.M.; Setlow, P. Bacillus megaterium spore protease: Synthesis and processing of precursor forms during sporulation and germination. J. Biol. Chem. 1982, 257, 10838–10845. [Google Scholar] [CrossRef]
- Setlow, P. Small acid-soluble, spore proteins of Bacillus species: Structure, synthesis, genetics, function and degradation. Annu. Rev. Microbiol. 1988, 42, 319–338. [Google Scholar] [CrossRef]
- Illades-Aguiar, B.; Setlow, P. Autoprocessing of the protease that degrades small, acid-soluble proteins of spores of Bacillus species is triggered by low pH, dehydration, and dipicolinic acid. J. Bacteriol. 1994, 176, 7032–7037. [Google Scholar] [CrossRef] [Green Version]
- Verhoef, J.; van Boven, C.P.A.; Holtrigter, B. Host controlled modification and restriction of phages in coagulase negative staphylococci. J. Gen. Microbiol. 1972, 71, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Coyette, J.; Ghuysen, J.M. Structure of the cell wall of Staphylococcus aureus, strain Copenhagen. IX. Teichoic acid and phage adsorption. Biochemistry 1968, 7, 2385–2389. [Google Scholar] [CrossRef]
- Holme, S.; Math, V.; Pitts, K.; Barnard, A.; Wang, H.; Wilkinson, A.; Fairhead, H. Low Propensity for S. aureus to Develop Resistance to SASP or its Delivery Vector. In Proceedings of the Interscience Conference Antimicrobe Agents Chemotherapy, Washington, DC, USA, 25–28 October 2008. Poster F1-4001. [Google Scholar]
- Duplessis, M.; Moineau, S. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol. Microbiol. 2001, 41, 325–336. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Pluckthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairhead, H.; Wilkinson, A. Modified bacteriophage. European Patent No. EP3340200, 24 February 2021. [Google Scholar]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- McCallin, S.; Sacher, J.C.; Zheng, J.; Chan, B.K. Current State of Compassionate Phage Therapy. Viruses 2019, 11, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails to Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mechanism of action of SASPject PT1.2 and SASP.

Figure 2.
Modification of phi11 genome (A) showing holin and amidase genes (B) and creation of SASPject PT1.2 by deletion of holin gene and promoter driving holin and amidase genes by insertion of SASP-C gene under the control of native S. aureus promoter, fbaA and, selectable marker, cadmium resistance gene (C). Phi11 genome visualized using CGView [21].
Figure 2.
Modification of phi11 genome (A) showing holin and amidase genes (B) and creation of SASPject PT1.2 by deletion of holin gene and promoter driving holin and amidase genes by insertion of SASP-C gene under the control of native S. aureus promoter, fbaA and, selectable marker, cadmium resistance gene (C). Phi11 genome visualized using CGView [21].

Figure 3.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed S. aureus MSSA/MRSA (strains 100085/88048 respectively) cultures. Viable cells counts are shown for the strain written in bold letters. Results of a representative experiment are shown.
Figure 3.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed S. aureus MSSA/MRSA (strains 100085/88048 respectively) cultures. Viable cells counts are shown for the strain written in bold letters. Results of a representative experiment are shown.

Figure 4.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed S. aureus MSSA/MRSA (strains 100085/17046 respectively) cultures. Viable cells counts are shown for the strain written in bold letters. Results of a representative experiment are shown.
Figure 4.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed S. aureus MSSA/MRSA (strains 100085/17046 respectively) cultures. Viable cells counts are shown for the strain written in bold letters. Results of a representative experiment are shown.

Figure 5.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed cultures of MRSA strain 88048 and S. epidermidis 7,020 (abbreviated to “S.e.” in the chart legend). Viable cell counts are shown for the strain written in bold letters. Results of a representative experiment are shown.
Figure 5.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed cultures of MRSA strain 88048 and S. epidermidis 7,020 (abbreviated to “S.e.” in the chart legend). Viable cell counts are shown for the strain written in bold letters. Results of a representative experiment are shown.

Figure 6.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed cultures of MRSA and S. haemolyticus 133034 (abbreviated to “S.h.” in the chart legend). Viable cell counts are shown for the strain written in bold letters. Results of a representative experiment are shown.
Figure 6.
Time-kill curve assay showing efficacy of SASPject PT1.2 in mixed cultures of MRSA and S. haemolyticus 133034 (abbreviated to “S.h.” in the chart legend). Viable cell counts are shown for the strain written in bold letters. Results of a representative experiment are shown.

Figure 7.
Time-kill curve assay of SASPject PT1.2 activity against S. aureus USA 300 over 1 h. Results of a representative experiment are shown.
Figure 7.
Time-kill curve assay of SASPject PT1.2 activity against S. aureus USA 300 over 1 h. Results of a representative experiment are shown.

Figure 8.
Time-kill curve analysis of SASPject PT1.2 activity, at concentrations ranging 106 to 108 pfu/mL, against EMRSA-15 strain 02ST4127 treated over 24 h. Data plotted are the means of 2 or 3 (107 pfu/mL only) replicates. Error bars represent 1 standard deviation.
Figure 8.
Time-kill curve analysis of SASPject PT1.2 activity, at concentrations ranging 106 to 108 pfu/mL, against EMRSA-15 strain 02ST4127 treated over 24 h. Data plotted are the means of 2 or 3 (107 pfu/mL only) replicates. Error bars represent 1 standard deviation.

Figure 9.
Time-kill curve analysis of SASPject PT1.2 activity against EMRSA-15 strain 02ST4127, at viable cell concentrations of 103, 105, and 107 cfu/mL, over 6 h. The means of duplicate samples are plotted. Error bars represent 1 standard deviation.
Figure 9.
Time-kill curve analysis of SASPject PT1.2 activity against EMRSA-15 strain 02ST4127, at viable cell concentrations of 103, 105, and 107 cfu/mL, over 6 h. The means of duplicate samples are plotted. Error bars represent 1 standard deviation.

Figure 10.
Time-kill curve assay showing the efficacy of SASPject PT1.2 against S. aureus MRSA USA300 and EMRSA-16 in the presence and absence of human serum albumin (HSA) at 50 g/L. Data points represent the mean of 2 replicates and error bars represent 1 standard deviation.
Figure 10.
Time-kill curve assay showing the efficacy of SASPject PT1.2 against S. aureus MRSA USA300 and EMRSA-16 in the presence and absence of human serum albumin (HSA) at 50 g/L. Data points represent the mean of 2 replicates and error bars represent 1 standard deviation.

Figure 11.
Assessment of resistance development in serially passaged EMRSA strain O2ST4285 by time-kill curve assay of cells from passage day 52 (3 and 6 × 104 pfu/mL passaged cultures) and passage day 46 (1.5 × 104 pfu/mL passaged cultures) exposed to SASPject PT1.2. Passaging was carried out in triplicate, and time-kill curve assays were carried out in singlicate on each of the triplicate cultures. Cells that were passaged in singlicate in the absence of PT1.2 for 52 days were exposed to PT1.2 as a control (Control). The mean number of viable cells for each passaging condition following time-kill curve assay with PT1.2 is plotted. Error bars for the passaged conditions represent 1 standard deviation.
Figure 11.
Assessment of resistance development in serially passaged EMRSA strain O2ST4285 by time-kill curve assay of cells from passage day 52 (3 and 6 × 104 pfu/mL passaged cultures) and passage day 46 (1.5 × 104 pfu/mL passaged cultures) exposed to SASPject PT1.2. Passaging was carried out in triplicate, and time-kill curve assays were carried out in singlicate on each of the triplicate cultures. Cells that were passaged in singlicate in the absence of PT1.2 for 52 days were exposed to PT1.2 as a control (Control). The mean number of viable cells for each passaging condition following time-kill curve assay with PT1.2 is plotted. Error bars for the passaged conditions represent 1 standard deviation.

Figure 12.
Assessment of resistance development in serially passaged MRSA USA300 by exposing passaged cells to PT1.2 at passage days 7, 14, and 21, and incubating with PT1.2 for 4 h before assessing viable cell count. Triplicate cultures were set up for each of the phage concentrations, and each culture was assessed via the kill experiment in singlicate. Results shown are the mean viable cell counts for the three cultures passaged with each PT1.2 concentration, following kill experiment analysis. Error bars represent 1 standard deviation. The control was MRSA USA300 simultaneously passaged for the same lengths of time in the absence of PT1.2, prior to assessment of viable cell count after exposure to PT1.2 for 4 h.
Figure 12.
Assessment of resistance development in serially passaged MRSA USA300 by exposing passaged cells to PT1.2 at passage days 7, 14, and 21, and incubating with PT1.2 for 4 h before assessing viable cell count. Triplicate cultures were set up for each of the phage concentrations, and each culture was assessed via the kill experiment in singlicate. Results shown are the mean viable cell counts for the three cultures passaged with each PT1.2 concentration, following kill experiment analysis. Error bars represent 1 standard deviation. The control was MRSA USA300 simultaneously passaged for the same lengths of time in the absence of PT1.2, prior to assessment of viable cell count after exposure to PT1.2 for 4 h.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Details of methicillin resistant S. aureus (MRSA) isolates (n = 187) and methicillin sensitive S. aureus (MSSA) isolates (n = 38) with log10 drop in viability at 3 h (3 h kill assay) or 4 h (time-kill curves).
Table 1.
Details of methicillin resistant S. aureus (MRSA) isolates (n = 187) and methicillin sensitive S. aureus (MSSA) isolates (n = 38) with log10 drop in viability at 3 h (3 h kill assay) or 4 h (time-kill curves).
| Character | Detail (Total No. of Isolates, n = 225) (Total No. of Isolates in the 3 h (3 h) or Time-Kill (tk) Assays) | No. of Isolates | |||||
|---|---|---|---|---|---|---|---|
| 3 h Kill Assay log10 Drop at 3 h | Time-Kill Curves log10 Drop at 4 h | ||||||
| ≥2 | ≥3 | ≥4 | ≥2 | ≥3 | ≥4 | ||
| mecA | mecA− (38) (3 h = 34; tk = 4) | 26 | 26 | 23 | 3 | 3 | 2 |
| mecA+ (187) (3 h = 163; tk = 24) | 158 | 156 | 153 | 22 | 21 | 15 | |
| Panton Valentine Leukocidin (PVL) | PVL− (3 h = 10) | 10 | 10 | 10 | |||
| PVL+ (3 h = 12) | 12 | 12 | 12 | ||||
| SCCmec | I (3 h = 22) | 21 | 21 | 20 | |||
| II (3 h = 31) | 31 | 31 | 31 | ||||
| III (3 h = 23) | 23 | 23 | 23 | ||||
| IV (3 h = 38) | 37 | 36 | 36 | ||||
| V (3 h = 1) | 1 | 1 | 1 | ||||
| Untypeable (3 h = 3) | 3 | 3 | 3 | ||||
| Sequence type | 1 (3 h = 3) | 3 | 3 | 3 | |||
| 22 (3 h = 15) | 15 | 15 | 15 | ||||
| 30 (3 h = 1) | 1 | 1 | 1 | ||||
| 36 (3 h = 12) | 12 | 12 | 12 | ||||
| 80 (3 h = 2) | 2 | 2 | 2 | ||||
| 239 (3 h = 6) | 6 | 6 | 6 | ||||
| 240 (3 h = 2) | 2 | 2 | 2 | ||||
| 247 (3 h = 4) | 4 | 4 | 4 | ||||
| 5 (3 h = 6) | 6 | 5 | 5 | ||||
| 8 (3 h = 16) | 16 | 15 | 15 | ||||
| USA type | ORSA-I (3 h = 7) | 7 | 7 | 7 | |||
| ORSA-II (3 h = 13) | 13 | 12 | 12 | ||||
| ORSA-III (3 h = 7) | 7 | 7 | 7 | ||||
| ORSA-IV (3 h = 14) | 14 | 14 | 14 | ||||
| ORSA-I/III/IV (3 h = 11) | 11 | 10 | 10 | ||||
| USA-100 (3 h = 1; tk = 1) | 1 | 0 | 0 | 1 | 1 | 1 | |
| USA-200 (13), -300 (1), -400 (1), -500 (1), -700 (1), -800 (1), -1000 (1), -1100 (1) (3 h = 20) | 12 | 12 | 12 | ||||
| VISA | VISA (3 h = 3; tk = 3) | 2 | 0 | 0 | 2 | 2 | 0 |
| hVISA | hVISA (3 h = 9; tk = 2) | 9 | 9 | 0 | 1 | 0 | 0 |
| Historic Clones | Archaic/Iberian (3 h = 15) | 15 | 14 | 14 | |||
| Brazilian/Hungarian (3 h = 28) | 28 | 28 | 28 | ||||
| Pediatric (3 h = 6) | 6 | 6 | 6 | ||||
| UK epidemic clones | EMRSA-1 (1), -2 (1), -3 (3), -4 (3), -5 (2), -6 (3), -7 (2), -8 (2), -9 (2), -10 (2), -11 (2), -12 (3) -13 (2), -14 (1), -15 (14), -16 (11), -17 (3) (3 h = 57) | 54 | 54 | 54 | |||
| EMRSA-6 (3 h = 3) | 3 | 3 | 2 | ||||
| Community MRSA | (3 h = 10; tk = 5) | 10 | 10 | 10 | 5 | 5 | 5 |
| Geographic location | Australia (3 h = 6) | 6 | 6 | 6 | |||
| Japan (3 h = 4) | 4 | 3 | 3 | ||||
| USA (3 h = 16) | 15 | 15 | 15 | ||||
| Denmark (3 h = 1) | 1 | 1 | 1 | ||||
| France (3 h = 2) | 2 | 2 | 2 | ||||
| Ireland (3 h = 5) | 5 | 5 | 5 | ||||
Table 2.
Log10 reduction in viable cells of coagulase negative Staphylococcal species and strains tested in 3 h kill assays.
Table 2.
Log10 reduction in viable cells of coagulase negative Staphylococcal species and strains tested in 3 h kill assays.
| Staphylococcal Species | Cambridge Identity Codes (CC) | Log10 Reduction in Viable Cells |
|---|---|---|
| S. epidermidis | 72003 | 0 |
| 72037 | 0 | |
| 72025 | 0 | |
| 72004 | 0 | |
| 72029 | 0 | |
| 72020 | 0 | |
| 72030 | 0 | |
| S. haemolyticus | 133072 | 0 |
| 133034 | 0 | |
| 133068 | 0 | |
| S. warneri | 133029 | 0 |
| 133019 | 0 | |
| S. hominis | 133075 | 0 |
| 133097 | 0 | |
| S. cohnii | 133089 | 0 |
| 133041 | 0 | |
| S. capitis | 133095 | 0 |
| 133092 | 0 | |
| S. simulans | 133080 | 0 |
| 133053 | 0 | |
| S. lugdunensis | 133074 | 0 |
| 133082 | 0 | |
| S. saprophyticus | 133002 | 0 |
| 133091 | 0 |
Table 3.
List of coagulase negative Staphylococcal species and strains tested for sensitivity against PT1.2 in 3 h kill assays.
Table 3.
List of coagulase negative Staphylococcal species and strains tested for sensitivity against PT1.2 in 3 h kill assays.
| Staphylococcal Species | Cambridge Identity Codes (CC) | Methicillin Sensitivity Profile |
|---|---|---|
| S. epidermidis | 72003 | S |
| 72037 | S | |
| 72025 | S | |
| 72004 | R | |
| 72029 | R | |
| 72020 | R | |
| 72030 | R | |
| S. haemolyticus | 133072 | S |
| 133034 | R | |
| 133068 | R | |
| S. warneri | 133029 | S |
| 133019 | R | |
| S. hominis | 133075 | S |
| 133097 | R | |
| S. cohnii | 133089 | S |
| 133041 | R | |
| S. capitis | 133095 | S |
| 133092 | R | |
| S. simulans | 133080 | S |
| 133053 | R | |
| S. lugdunensis | 133074 | S |
| 133082 | R | |
| S. saprophyticus | 133002 | S |
| 133091 | Unknown |
Table 4.
List of Staphylococcal isolates used in mixed bacterial culture experiments.
| Staphylococcal Species | Cambridge Identity Codes (CC) | Methicillin Sensitivity Profile |
|---|---|---|
| S. aureus | 88048 | R |
| 17046 | R | |
| 100085 | S | |
| S. epidermidis | 72020 | R |
| S. haemolyticus | 133034 | R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cass, J.; Barnard, A.; Fairhead, H. Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP. Pharmaceuticals 2021, 14, 1038. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101038
AMA Style
Cass J, Barnard A, Fairhead H. Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP. Pharmaceuticals. 2021; 14(10):1038. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101038
Chicago/Turabian StyleCass, James, Anne Barnard, and Heather Fairhead. 2021. "Engineered Bacteriophage as a Delivery Vehicle for Antibacterial Protein, SASP" Pharmaceuticals 14, no. 10: 1038. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101038
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.