
Studies of Potency and Efficacy of an Optimized Artemisinin-Quinoline Hybrid against Multiple Stages of the Plasmodium Life Cycle

 ,
,  , , , , ,
, , , , ,  ,
,  and
and 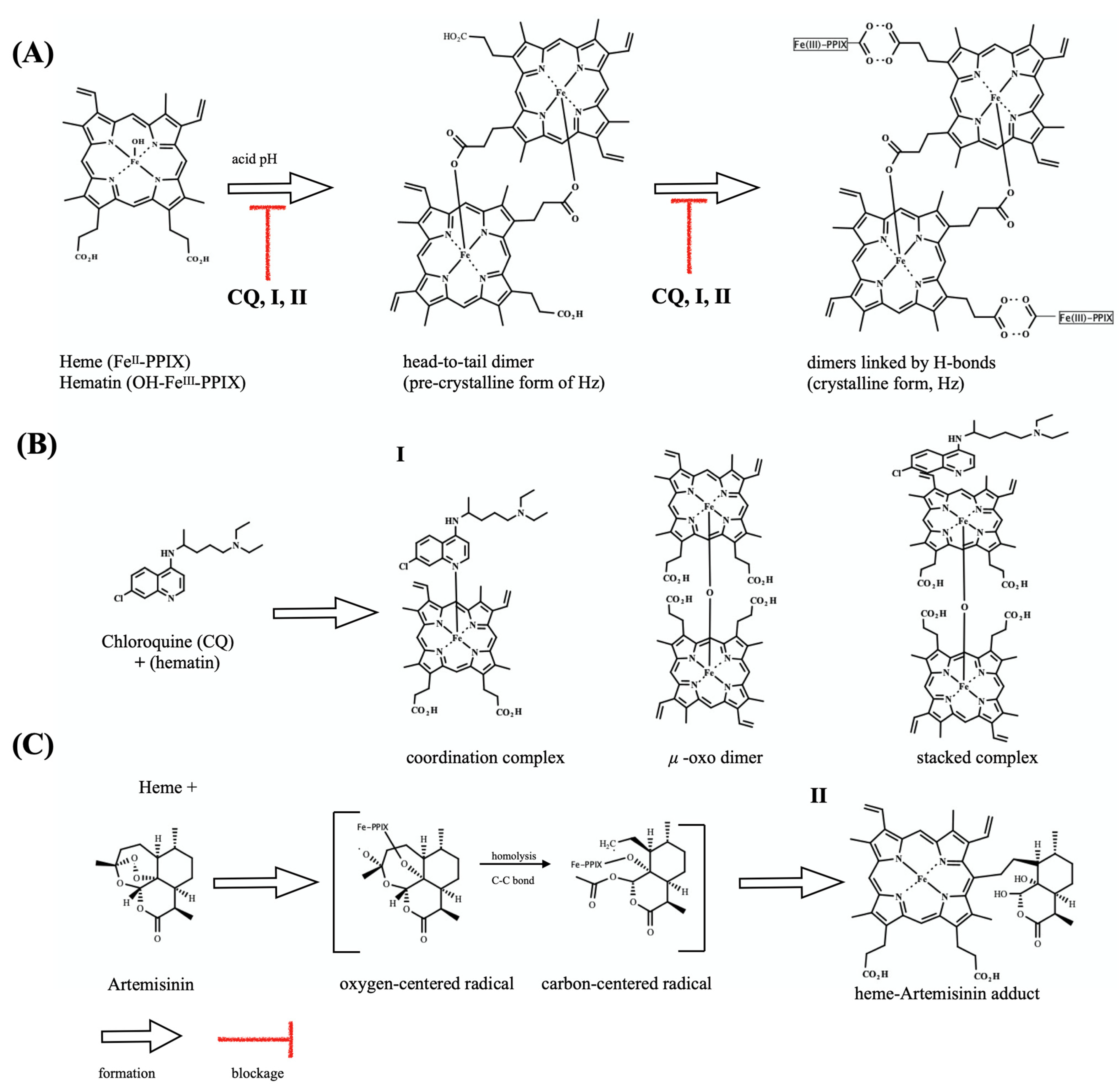{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
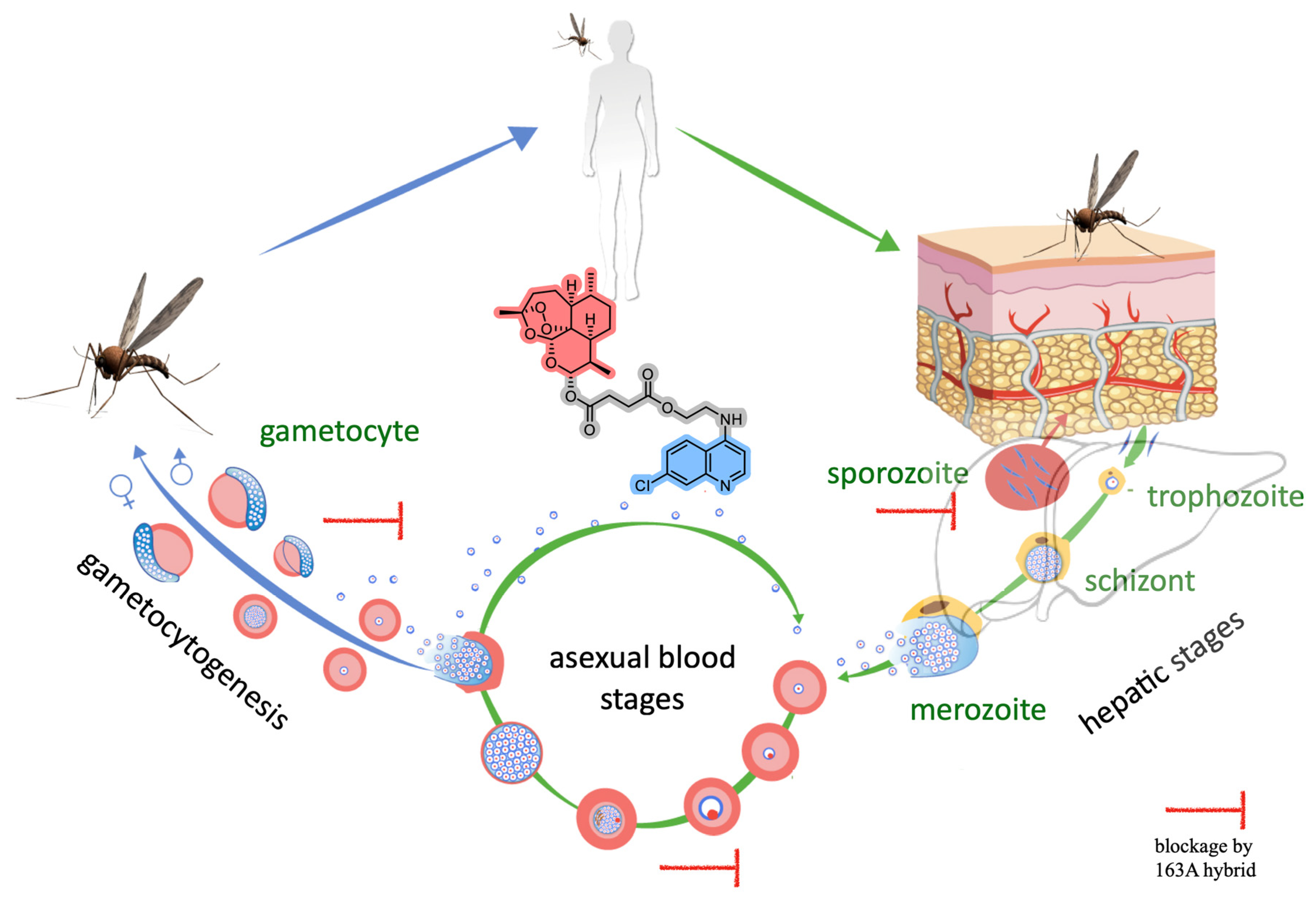{kind=link}
Abstract
:1. Introduction
2. Results
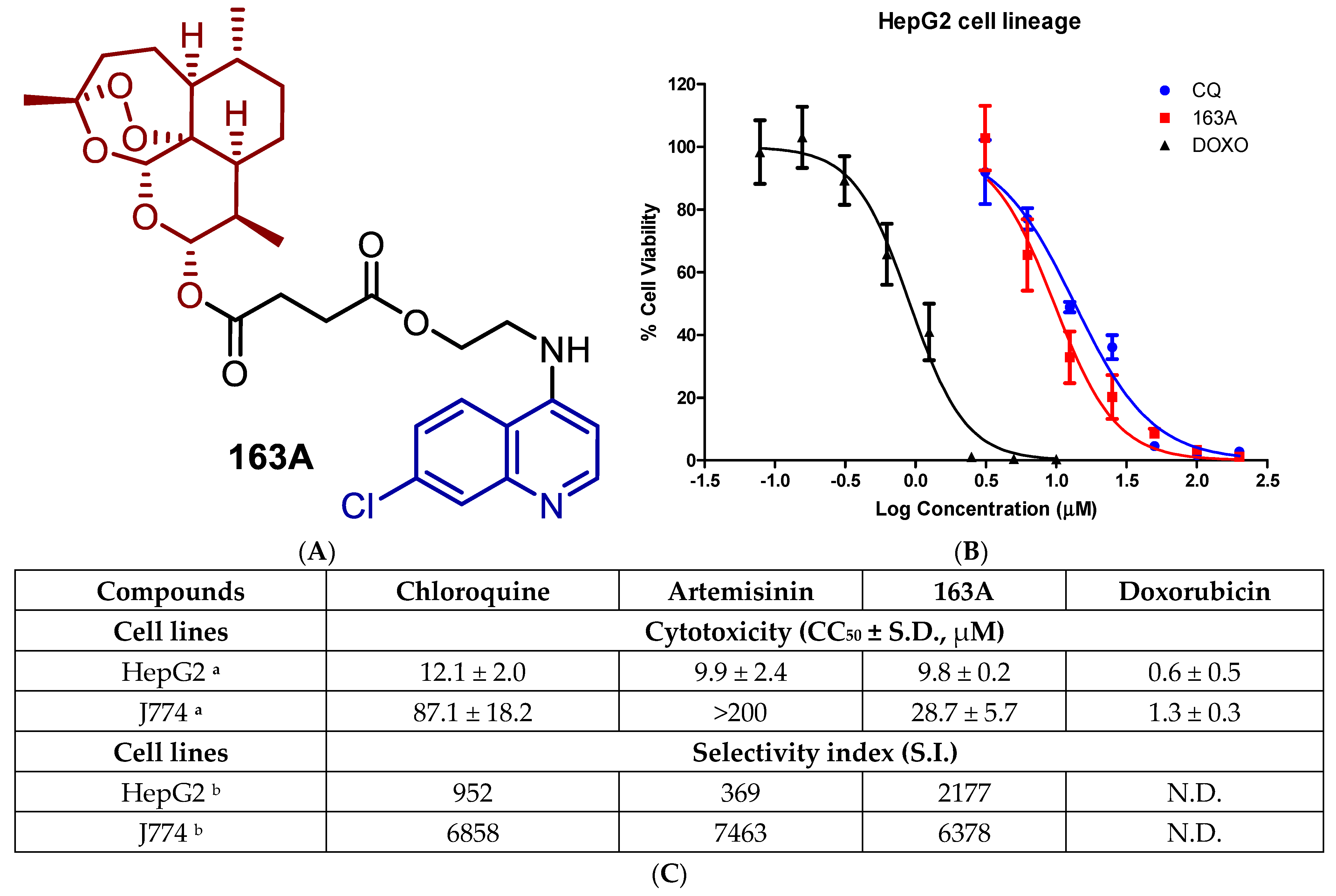
2.1. Toxicity for Host Cells and Selectivity
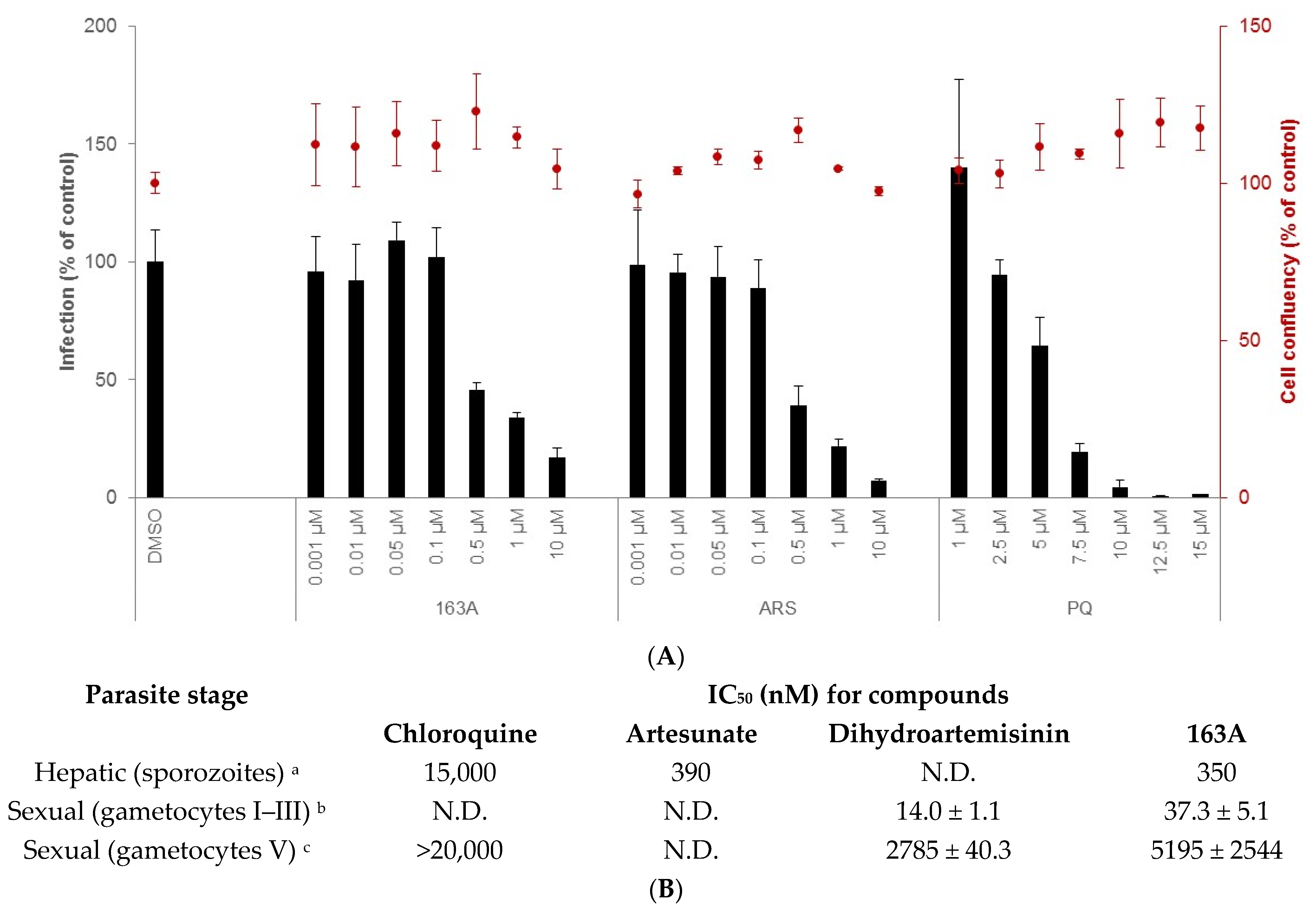
2.2. Inhibition on Hepatic and Sexual Stages
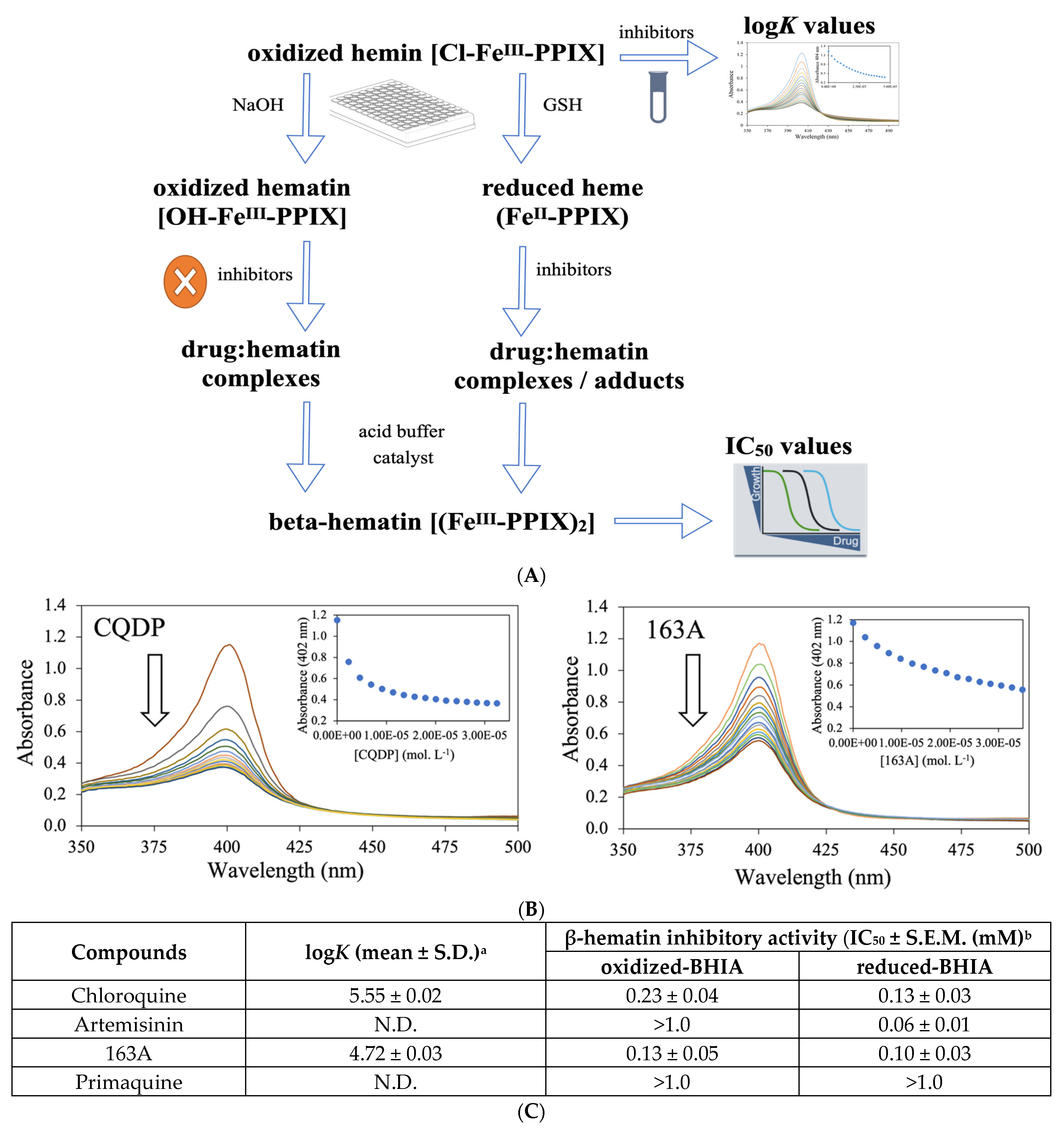
2.3. Dual Role on the Heme Detoxification Process
2.4. Parasitemia Profile in P. berghei-Infected Mice
3. Discussion
4. Materials and Methods
4.1. General Materials
4.2. Cytotoxicity Assay in Mammalian Cells
4.3. P. berghei Liver Stage
4.4. P. falciparum Gametocytes
4.5. β-Hematin Inhibition Activity (BHIA)
4.6. Determination of the Association Constant to Ferriprotoporphyrin IX
4.7. Mice
4.8. Parasites
4.9. P. berghei-Infected Mice (Thompson Test)
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tam, G.; Cowling, B.J.; Maude, R.J. Analysing human population movement data for malaria control and elimination. Malar. J. 2021, 20, 294. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, S.W.; Thomas, M.B.; Kleinschmidt, I. Threats to the effectiveness of insecticide-treated bednets for malaria control: Thinking beyond insecticide resistance. Lancet Glob. Health 2021, 9, e1325–e1331. [Google Scholar] [CrossRef]
- Hughes, E.; Wallender, E.; Ali, A.M.; Jagannathan, P.; Savic, R.M. Malaria PK/PD and the Role Pharmacometrics Can Play in the Global Health Arena: Malaria Treatment Regimens for Vulnerable Populations. Clin. Pharm. Ther. 2021, 110, 926–940. [Google Scholar] [CrossRef]
- Moxon, C.A.; Gibbins, M.; McGuinness, D.; Milner, D.A.; Marti, M. New Insights into Malaria Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 315–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffey, M.; Blasco, B.; Burrows, J.N.; Wells, T.N.; Fidock, D.A.; Leroy, D. Assessing risks of Plasmodium falciparum resistance to select next-generation antimalarials. Trends Parasitol. 2021, 37, 709–721. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, K.A.; Egan, T.J. Heme Detoxification in the Malaria Parasite: A Target for Antimalarial Drug Development. Accounts Chem. Res. 2021, 54, 2649–2659. [Google Scholar] [CrossRef]
- Kapishnikov, S.; Hempelmann, E.; Elbaum, M.; Als-Nielsen, J.; Leiserowitz, L. Malaria Pigment Crystals: The Achilles′ Heel of the Malaria Parasite. ChemMedChem 2021, 16, 1515–1532. [Google Scholar] [CrossRef]
- Hanscheid, T.; Egan, T.J.; Grobusch, M.P. Haemozoin: From melatonin pigment to drug target, diagnostic tool, and immune modulator. Lancet Infect. Dis. 2007, 7, 675–685. [Google Scholar] [CrossRef]
- Openshaw, R.; Maepa, K.; Benjamin, S.J.; Wainwright, L.; Combrinck, J.M.; Hunter, R.; Egan, T.J. A Diverse Range of Hemozoin Inhibiting Scaffolds Act on Plasmodium falciparum as Heme Complexes. ACS Infect. Dis. 2021, 7, 362–376. [Google Scholar] [CrossRef]
- Olafson, K.N.; Nguyen, T.Q.; Rimer, J.D.; Vekilov, P.G. Antimalarials inhibit hematin crystallization by unique drug–surface site interactions. Proc. Natl. Acad. Sci. USA 2017, 114, 7531–7536. [Google Scholar] [CrossRef] [Green Version]
- Garah, F.B.-E.; Wong, M.H.-L.; Amewu, R.K.; Muangnoicharoen, S.; Maggs, J.L.; Stigliani, J.-L.; Park, B.K.; Chadwick, J.; Ward, S.A.; O’Neill, P.M. Comparison of the Reactivity of Antimalarial 1,2,4,5-Tetraoxanes with 1,2,4-Trioxolanes in the Presence of Ferrous Iron Salts, Heme, and Ferrous Iron Salts/Phosphatidylcholine. J. Med. Chem. 2011, 54, 6443–6455. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Charman, W.N.; Chiu, F.C.K.; Prankerd, R.J.; Dong, Y.; Vennerstrom, J.L.; Charman, S.A. Relationship between Antimalarial Activity and Heme Alkylation for Spiro- and Dispiro-1,2,4-Trioxolane Antimalarials. Antimicrob. Agents Chemother. 2008, 52, 1291–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loup, C.; Lelièvre, J.; Benoit-Vical, F.; Meunier, B. Trioxaquines and Heme-Artemisinin Adducts Inhibit the In Vitro Formation of Hemozoin Better than Chloroquine. Antimicrob. Agents Chemother. 2007, 51, 3768–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, C.-J.; Ni Chia, W.; Loh, C.C.Y.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.-X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef]
- Ma, W.; Balta, V.A.; West, R.; Newlin, K.N.; Miljanić, O.Š.; Sullivan, D.J.; Vekilov, P.G.; Rimer, J.D. A second mechanism employed by artemisinins to suppress Plasmodium falciparum hinges on inhibition of hematin crystallization. J. Biol. Chem. 2021, 296, 100123. [Google Scholar] [CrossRef]
- Van der Pluijm, R.W.; Tripura, R.; Hoglund, R.M.; Phyo, A.P.; Lek, D.; Islam, A.U.; Anvikar, A.R.; Satpathi, P.; Satpathi, S.; Behera, P.K.; et al. Triple artemisinin-based combination therapies versus artemisinin-based combination therapies for uncomplicated Plasmodium falciparum malaria: A multicentre, open-label, randomised clinical trial. Lancet 2020, 395, 1345–1360. [Google Scholar] [CrossRef]
- Wang, J.; Xu, C.; Liao, F.L.; Jiang, T.; Krishna, S.; Tu, Y. A Temporizing Solution to “Artemisinin Resistance”. N. Engl. J. Med. 2019, 380, 2087–2089. [Google Scholar] [CrossRef]
- Santos-Magalhães, N.S.; Mosqueira, V.C.F. Nanotechnology applied to the treatment of malaria. Adv. Drug Deliv. Rev. 2010, 62, 560–575. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, T.; Çapcı Karagöz, A.; Reiter, C.; Tsogoeva, S.B. Artemisinin-Derived Dimers: Potent Antimalarial and Anticancer Agents. J. Med. Chem. 2016, 59, 7360–7388. [Google Scholar] [CrossRef]
- Çapcı, A.; Herrmann, L.; Kumar, H.M.S.; Fröhlich, T.; Tsogoeva, S.B. Artemisinin-derived dimers from a chemical perspective. Med. Res. Rev. 2021, 41, 2927–2970. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Progress in the Development of Synthetic Hybrids of Natural or Unnatural Bioactive Compounds for Medicinal Chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef]
- Walsh, J.; Bell, A. Hybrid Drugs for Malaria. Curr. Pharm. Des. 2009, 15, 2970–2985. [Google Scholar] [CrossRef]
- Benoit-Vical, F.; Lelièvre, J.; Berry, A.; Deymier, C.; Dechy-Cabaret, O.; Cazelles, J.; Loup, C.; Robert, A.; Magnaval, J.-F.; Meunier, B. Trioxaquines Are New Antimalarial Agents Active on All Erythrocytic Forms, Including Gametocytes. Antimicrob. Agents Chemother. 2007, 51, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Coslédan, F.; Fraisse, L.; Pellet, A.; Guillou, F.; Mordmüller, B.; Kremsner, P.G.; Moreno, A.; Mazier, D.; Maffrand, J.-P.; Meunier, B. Selection of a trioxaquine as an antimalarial drug candidate. Proc. Natl. Acad. Sci. USA 2008, 105, 17579–17584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwedel, C.; Tsogoeva, S.B.; Wei, S.; Efferth, T. Cytotoxicity of Artesunic Acid Homo- and Heterodimer Molecules toward Sensitive and Multidrug-Resistant CCRF-CEM Leukemia Cells. J. Med. Chem. 2010, 53, 4842–4848. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.-S.; Guantai, E.; Nell, M.; van Rensburg, C.E.; Ncokazi, K.; Egan, T.J.; Hoppe, H.C.; Chibale, K. Effects of highly active novel artemisinin–chloroquinoline hybrid compounds on β-hematin formation, parasite morphology and endocytosis in Plasmodium falciparum. Biochem. Pharmacol. 2011, 82, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Hahn, F.; Belmudes, L.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Couté, Y.; Marschall, M.; Tsogoeva, S.B. Synthesis of Artemisinin-Derived Dimers, Trimers and Dendrimers: Investigation of Their Antimalarial and Antiviral Activities Including Putative Mechanisms of Action. Chemistry 2018, 24, 8103–8113. [Google Scholar] [CrossRef]
- Zhan, W.; Liu, Y.J.; Yang, C.; Zhang, H.; Harris, J.C.; Wang, R.; Zhu, S.; Sherman, J.; Sukenick, G.; Rodriguez, A.; et al. Artemisinin-based hybrids produce intracellular proteasome inhibitors that overcome resistance in Plasmodium falciparum. bioRxiv 2021. [Google Scholar] [CrossRef]
- Çapcı, A.; Lorion, M.M.; Wang, H.; Simon, N.; Leidenberger, M.; Silva, M.C.B.; Moreira, D.R.M.; Zhu, Y.; Meng, Y.; Chen, J.Y.; et al. Artemisinin–(Iso)quinoline Hybrids by C−H Activation and Click Chemistry: Combating Multidrug-Resistant Malaria. Angew. Chem. Int. Ed. 2019, 58, 13066–13079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontinha, D.; Moules, I.; Prudêncio, M. Repurposing Drugs to Fight Hepatic Malaria Parasites. Molecules 2020, 25, 3409. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Anti-malarial drug effects on parasite dynamics in vivax malaria. Malar. J. 2021, 20, 161. [Google Scholar] [CrossRef]
- Ngotho, P.; Soares, A.B.; Hentzschel, F.; Achcar, F.; Bertuccini, L.; Marti, M. Revisiting gametocyte biology in malaria parasites. FEMS Microbiol. Rev. 2019, 43, 401–414. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Benoit-Vical, F. Effects of Antimalarial Molecules on the Gametocyte Stage of Plasmodium falciparum: The Debate. J. Med. Chem. 2012, 55, 10328–10344. [Google Scholar] [CrossRef]
- Ploemen, I.H.J.; Prudêncio, M.; Douradinha, B.; Ramesar, J.; Fonager, J.; Van Gemert, G.-J.; Luty, A.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G.; et al. Visualisation and Quantitative Analysis of the Rodent Malaria Liver Stage by Real Time Imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, S.; Silvestrini, F.; Dechering, K.; Corbett, Y.; Parapini, S.; Timmerman, M.; Galastri, L.; Basilico, N.; Sauerwein, R.; Alano, P.; et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J. Antimicrob. Chemother. 2013, 68, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Capela, R.; Cabal, G.; Rosenthal, P.J.; Gut, J.; Mota, M.M.; Moreira, R.; Lopes, F.; Prudêncio, M. Design and Evaluation of Primaquine-Artemisinin Hybrids as a Multistage Antimalarial Strategy. Antimicrob. Agents Chemother. 2011, 55, 4698–4706. [Google Scholar] [CrossRef] [PubMed]
- Delves, M.; Plouffe, D.; Scheurer, C.; Meister, S.; Wittlin, S.; Winzeler, E.; Sinden, R.E.; Leroy, D. The Activities of Current Antimalarial Drugs on the Life Cycle Stages of Plasmodium: A Comparative Study with Human and Rodent Parasites. PLoS Med. 2012, 9, e1001169. [Google Scholar] [CrossRef] [Green Version]
- Parapini, S.; Olliaro, P.; Navaratnam, V.; Taramelli, D.; Basilico, N. Stability of the Antimalarial Drug Dihydroartemisinin under Physiologically Relevant Conditions: Implications for Clinical Treatment and Pharmacokinetic and In Vitro Assays. Antimicrob. Agents Chemother. 2015, 59, 4046–4052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, T.S.; Villarreal, W.; Couto, C.C.; Moreira, D.R.M.; Navarro, M.; Machado, M.; Prudêncio, M.; Batista, A.A.; Soares, M.B.P. Platinum(II)–chloroquine complexes are antimalarial agents against blood and liver stages by impairing mitochondrial function. Metallomics 2017, 9, 1548–1561. [Google Scholar] [CrossRef]
- Ribbiso, K.A.; Heller, L.E.; Taye, A.; Julian, E.; Willems, A.V.; Roepe, P.D. Artemisinin-Based Drugs Target the Plasmodium falciparum Heme Detoxification Pathway. Antimicrob. Agents Chemother. 2021, 65, e02137-20. [Google Scholar] [CrossRef] [PubMed]
- Egan, T.J.; Mavuso, W.W.; Ross, D.C.; Marques, H. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J. Inorg. Biochem. 1997, 68, 137–145. [Google Scholar] [CrossRef]
- Olafson, K.N.; Nguyen, T.Q.; Vekilov, P.G.; Rimer, J.D. Deconstructing Quinoline-Class Antimalarials to Identify Fundamental Physicochemical Properties of Beta-Hematin Crystal Growth Inhibitors. Chemistry 2017, 23, 13638–13647. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.R.; Page-Sharp, M.; Stoney, J.R.; Ilett, K.F.; Jago, J.D.; Batty, K.T. Pharmacokinetics, Pharmacodynamics, and Allometric Scaling of Chloroquine in a Murine Malaria Model. Antimicrob. Agents Chemother. 2011, 55, 3899–3907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birrell, G.W.; Chavchich, M.; Ager, A.L.; Shieh, H.-M.; Heffernan, G.D.; Zhao, W.; Krasucki, P.E.; Saionz, K.W.; Terpinski, J.; Schiehser, G.A.; et al. JPC-2997, a New Aminomethylphenol with High In Vitro and In Vivo Antimalarial Activities against Blood Stages of Plasmodium. Antimicrob. Agents Chemother. 2015, 59, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarda, G.; Jirawatcharadech, P.; Priestley, R.S.; Saif, A.; March, S.; Wong, M.H.L.; Leung, S.; Miller, A.B.; Baker, D.A.; Alano, P.; et al. Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat. Commun. 2019, 10, 3226. [Google Scholar] [CrossRef]
- Adjalley, S.H.; Johnston, G.L.; Li, T.; Eastman, R.T.; Ekland, E.H.; Eappen, A.G.; Richman, A.; Sim, B.K.L.; Lee, M.C.S.; Hoffman, S.L.; et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc. Natl. Acad. Sci. USA 2011, 108, E1214–E1223. [Google Scholar] [CrossRef] [Green Version]
- Lelièvre, J.; Almela, M.J.; Lozano, S.; Miguel, C.; Franco, V.; Leroy, D.; Herreros, E. Activity of Clinically Relevant Antimalarial Drugs on Plasmodium falciparum Mature Gametocytes in an ATP Bioluminescence “Transmission Blocking” Assay. PLoS ONE 2012, 7, e35019. [Google Scholar] [CrossRef] [Green Version]
- Newton, P.; van Vugt, M.; Teja-Isavadharm, P.; Siriyanonda, D.; Rasameesoroj, M.; Teerapong, P.; Ruangveerayuth, R.; Slight, T.; Nosten, F.; Suputtamongkol, Y.; et al. Comparison of Oral Artesunate and Dihydroartemisinin Antimalarial Bioavailabilities in Acute Falciparum Malaria. Antimicrob. Agents Chemother. 2002, 46, 1125–1127. [Google Scholar] [CrossRef] [Green Version]
- Lichorowic, C.L.; Zhao, Y.; Maher, S.P.; Padín-Irizarry, V.; Mendiola, V.C.; de Castro, S.T.; Worden, J.A.; Casandra, D.; Kyle, D.E.; Manetsch, R. Synthesis of Mono- and Bisperoxide-Bridged Artemisinin Dimers to Elucidate the Contribution of Dimerization to Antimalarial Activity. ACS Infect. Dis. 2021, 7, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ncokazi, K.K.; Egan, T.J. A colorimetric high-throughput β-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal. Biochem. 2005, 338, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, B.A.; Mota, M.M.; Sultan, A.A.; Carvalho, L.H. Plasmodium berghei parasite transformed with green fluorescent protein for screening blood schizontocidal agents. Int. J. Parasitol. 2004, 34, 485–490. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quadros, H.C.; Çapcı, A.; Herrmann, L.; D’Alessandro, S.; Fontinha, D.; Azevedo, R.; Villarreal, W.; Basilico, N.; Prudêncio, M.; Tsogoeva, S.B.; et al. Studies of Potency and Efficacy of an Optimized Artemisinin-Quinoline Hybrid against Multiple Stages of the Plasmodium Life Cycle. Pharmaceuticals 2021, 14, 1129. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111129
Quadros HC, Çapcı A, Herrmann L, D’Alessandro S, Fontinha D, Azevedo R, Villarreal W, Basilico N, Prudêncio M, Tsogoeva SB, et al. Studies of Potency and Efficacy of an Optimized Artemisinin-Quinoline Hybrid against Multiple Stages of the Plasmodium Life Cycle. Pharmaceuticals. 2021; 14(11):1129. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111129
Chicago/Turabian StyleQuadros, Helenita C., Aysun Çapcı, Lars Herrmann, Sarah D’Alessandro, Diana Fontinha, Raquel Azevedo, Wilmer Villarreal, Nicoletta Basilico, Miguel Prudêncio, Svetlana B. Tsogoeva, and et al. 2021. "Studies of Potency and Efficacy of an Optimized Artemisinin-Quinoline Hybrid against Multiple Stages of the Plasmodium Life Cycle" Pharmaceuticals 14, no. 11: 1129. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111129