
The Cholecystokinin Type 2 Receptor, a Pharmacological Target for Pain Management

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. CCK2R Structure and Functions
2.1. Gene Localization and Related Diseases
2.2. Structural Features of the CCK2R
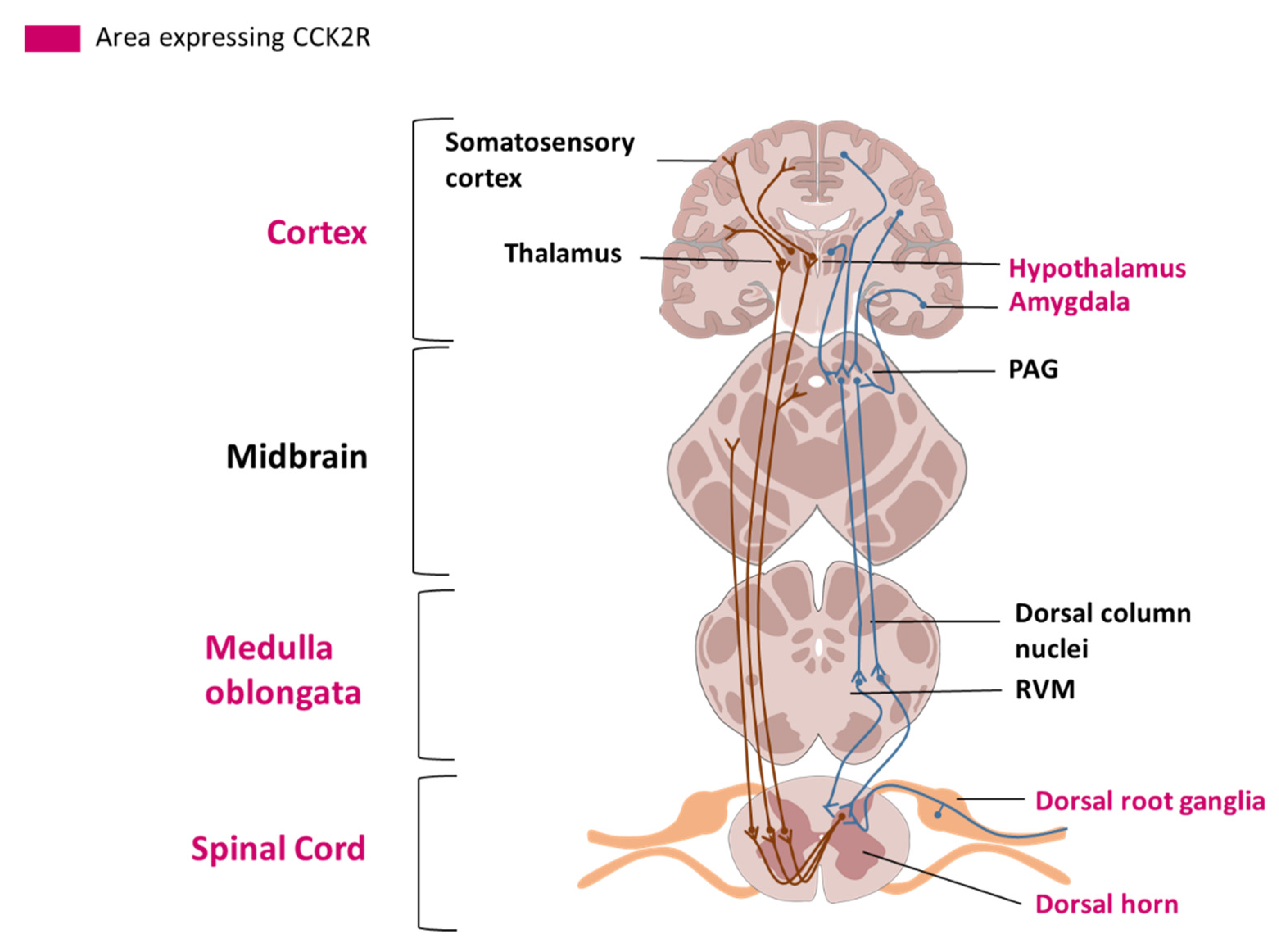
2.3. Cellular Localization and Tissue Distribution in the Nervous System
3. Involvement of CCK2R in Pain Modulation
3.1. The Heterodimerization of CCK2R and the Opioid Receptor Reduces Opioid Efficacy
3.2. CCK/CCK2R Facilitates Glutamate Release
3.3. CCK/CCK2R Increases Dopamine
3.4. CCK/CCK2R Decreases GABA Release
3.5. CCK/CCK2R Decreases K+ Channel Currents
4. Development of Pharmacological Modulators
5. Pharmacological Modulation of CCK2R in Preclinical Models of Pain
5.1. Model of Traumatic Injury at the Central Nervous System Level
5.2. Model of Traumatic Injury at the Peripheral Nervous System Level
5.3. Model of Burn Pain
5.4. Model of Diabetic Neuropathy
6. Clinical Relevance of CCK2R Pharmacological Targeting
6.1. Placebo
6.2. Postoperative Pain
6.3. Cancer Pain
6.4. Neuropathic Pain
7. Conclusions/Critical Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ivy, A.C.; Oldberg, E. A Hormone Mechanism for Gall-Bladder Contraction and Evacuation. Am. J. Physiol. Leg. Content 1928, 86, 599–613. [Google Scholar] [CrossRef]
- Wank, S.A. Cholecystokinin Receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 1995, 269, G628–G646. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Ahrén, B. CCKA Receptor Antagonism Inhibits Mechanisms Underlying CCK-8-Stimulated Insulin Release in Isolated Rat Islets. Eur. J. Pharmacol. 1991, 202, 253–257. [Google Scholar] [CrossRef]
- Rossetti, L.; Shulman, G.I.; Zawalich, W.S. Physiological Role of Cholecystokinin in Meal-Induced Insulin Secretion in Conscious Rats. Studies with L 364718, a Specific Inhibitor of CCK-Receptor Binding. Diabetes 1987, 36, 1212–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grider, J.R. Role of Cholecystokinin in the Regulation of Gastrointestinal Motility. J. Nutr. 1994, 124, 1334S–1339S. [Google Scholar] [CrossRef] [Green Version]
- Dufresne, M.; Seva, C.; Fourmy, D. Cholecystokinin and Gastrin Receptors. Physiol. Rev. 2006, 86, 805–847. [Google Scholar] [CrossRef] [Green Version]
- McRoberts, J.W. Cholecystokinin and Pain. Anesth. Prog. 1986, 33, 87–90. [Google Scholar]
- Vanderhaeghen, J.J.; Signeau, J.C.; Gepts, W. New Peptide in the Vertebrate CNS Reacting with Antigastrin Antibodies. Nature 1975, 257, 604–605. [Google Scholar] [CrossRef]
- Jackson, D.L.; Graff, C.B.; Richardson, J.D.; Hargreaves, K.M. Glutamate Participates in the Peripheral Modulation of Thermal Hyperalgesia in Rats. Eur. J. Pharmacol. 1995, 284, 321–325. [Google Scholar] [CrossRef]
- Léna, I.; Dhôtel, H.; Garbay, C.; Daugé, V. Involvement of D 2 Dopamine Receptors in the Opposing Effects of Two CCK-B Agonists in a Spatial Recognition Memory Task: Role of the Anterior Nucleus Accumbens. Psychopharmacology 2001, 153, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Q.; He, Q.-H.; Han, J.; Su, L.; Wan, Y. Heteromerization of μ-Opioid Receptor and Cholecystokinin B Receptor through the Third Transmembrane Domain of the μ-Opioid Receptor Contributes to the Anti-Opioid Effects of Cholecystokinin Octapeptide. Exp. Mol. Med. 2018, 50, 64. [Google Scholar] [CrossRef] [Green Version]
- Kamei, J.; Zushida, K. The Role of Spinal Cholecystokinin B Receptors in Thermal Allodynia and Hyperalgesia in Diabetic Mice. Brain Res. 2001, 892, 370–375. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.H.; Kim, Y.; Cho, H.; Hong, S.K.; Yoon, Y.W. Role of Spinal Cholecystokinin in Neuropathic Pain after Spinal Cord Hemisection in Rats. Neurosci. Lett. 2009, 462, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Noble, F.; Roques, B.P. CCK-B Receptor: Chemistry, Molecular Biology, Biochemistry and Pharmacology. Prog. Neurobiol. 1999, 58, 349–379. [Google Scholar] [CrossRef]
- Willard, M.D.; Lajiness, M.E.; Wulur, I.H.; Feng, B.; Swearingen, M.L.; Uhlik, M.T.; Kinzler, K.W.; Velculescu, V.E.; Sjöblom, T.; Markowitz, S.D.; et al. Somatic Mutations in CCK2R Alter Receptor Activity That Promote Oncogenic Phenotypes. Mol. Cancer Res. 2012, 10, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.J.; Gao, F. Structural Basis of Cholecystokinin Receptor Binding and Regulation. Pharm. Ther. 2008, 119, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piiper, A.; Stryjek-Kaminska, D.; Klengel, R.; Zeuzem, S. CCK, Carbachol, and Bombesin Activate Distinct PLC-Beta Isoenzymes via Gq/11 in Rat Pancreatic Acinar Membranes. Am. J. Physiol. 1997, 272, G135–G140. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-G.; Cong, B.; Li, Q.-X.; Chen, H.-Y.; Qin, J.; Fu, L.-H. Cholecystokinin Octapeptide Regulates Lipopolysaccharide-Activated B Cells Co-Stimulatory Molecule Expression and Cytokines Production In Vitro. Immunopharmacol. Immunotoxicol. 2011, 33, 157–163. [Google Scholar] [CrossRef]
- Stone, L.S.; Molliver, D.C. In Search of Analgesia: Emerging Poles of GPCRs in Pain. Mol. Interv. 2009, 9, 234–251. [Google Scholar] [CrossRef] [Green Version]
- Danigo, A.; Rovini, A.; Bessaguet, F.; Bouchenaki, H.; Bernard, A.; Sturtz, F.; Bourthoumieu, S.; Desmoulière, A.; Magy, L.; Demiot, C. The Angiotensin II Type 2 Receptor, a Target for Protection and Regeneration of the Peripheral Nervous System? Pharmaceuticals 2021, 14, 175. [Google Scholar] [CrossRef]
- Pohl, M.; Silvente-Poirot, S.; Pisegna, J.R.; Tarasova, N.I.; Wank, S.A. Ligand-Induced Internalization of Cholecystokinin Receptors. J. Biol. Chem. 1997, 272, 18179–18184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietl, M.M.; Probst, A.; Palacios, J.M. On the Distribution of Cholecystokinin Receptor Binding Sites in the Human Brain: An Autoradiographic Study. Synapse 1987, 1, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Gaudreau, P.; St-Pierre, S.; Pert, C.B.; Quirion, R. Cholecystokinin Receptors in Mammalian Brain: A Comparative Characterization and Visualization. Ann. N. Y. Acad. Sci. 1985, 448, 198–219. [Google Scholar] [CrossRef] [PubMed]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An Atlas of the Protein-Coding Genes in the Human, Pig, and Mouse Brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Dafny, N.; Dong, W.Q.; Prieto-Gomez, C.; Reyes-Vazquez, C.; Stanford, J.; Qiao, J.T. Lateral Hypothalamus: Site Involved in Pain Modulation. Neuroscience 1996, 70, 449–460. [Google Scholar] [CrossRef]
- Chudler, E.H.; Dong, W.K. The Role of the Basal Ganglia in Nociception and Pain. Pain 1995, 60, 3–38. [Google Scholar] [CrossRef]
- Neugebauer, V. 15. Amygdala Pain Mechanisms. Handb Exp. Pharm. 2015, 227, 261–284. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Dagerlind, A.; Elde, R.P.; Castel, M.N.; Broberger, C.; Wiesenfeld-Hallin, Z.; Hökfelt, T. Marked Increase in Cholecystokinin B Receptor Messenger RNA Levels in Rat Dorsal Root Ganglia after Peripheral Axotomy. Neuroscience 1993, 57, 227–233. [Google Scholar] [CrossRef]
- Ghilardi, J.R.; Allen, C.J.; Vigna, S.R.; McVey, D.C.; Mantyh, P.W. Trigeminal and Dorsal Root Ganglion Neurons Express CCK Receptor Binding Sites in the Rat, Rabbit, and Monkey: Possible Site of Opiate-CCK Analgesic Interactions. J. Neurosci. 1992, 12, 4854–4866. [Google Scholar] [CrossRef] [Green Version]
- Faris, P.; Komisaruk, B.; Watkins, L.; Mayer, D. Evidence for the Neuropeptide Cholecystokinin as an Antagonist of Opiate Analgesia. Science 1983, 219, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. Opioids, Sensory Systems and Chronic Pain. Eur. J. Pharmacol. 2013, 716, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Torres-López, J.E.; Juárez-Rojop, I.E.; Granados-Soto, V.; Diaz-Zagoya, J.C.; Flores-Murrieta, F.J.; Ortíz-López, J.U.S.; Cruz-Vera, J. Peripheral Participation of Cholecystokinin in the Morphine-Induced Peripheral Antinociceptive Effect in Non-Diabetic and Diabetic Rats. Neuropharmacology 2007, 52, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Wang, X.J.; Han, J.S. Cholecystokinin octapeptide (CCK-8) antagonized analgesia mediated by mu and kappa opioid receptors. Sheng Li Xue Bao 1990, 42, 219–225. [Google Scholar]
- Dourish, C.T.; O’Neill, M.F.; Coughlan, J.; Kitchener, S.J.; Hawley, D.; Iversen, S.D. The Selective CCK-B Receptor Antagonist L-365,260 Enhances Morphine Analgesia and Prevents Morphine Tolerance in the Rat. Eur. J. Pharmacol. 1990, 176, 35–44. [Google Scholar] [CrossRef]
- Gall, C.; Berry, L.M.; Hodgson, L.A. Cholecystokinin in the Mouse Hippocampus: Localization in the Mossy Fiber and Dentate Commissural Systems. Exp. Brain Res. 1986, 62, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Bradwejn, J.; Vasar, E. Cholecystokinin and Anxiety: From Neuron to Behavior; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 978-3-662-21705-4. [Google Scholar]
- Deng, P.-Y.; Xiao, Z.; Jha, A.; Ramonet, D.; Matsui, T.; Leitges, M.; Shin, H.-S.; Porter, J.E.; Geiger, J.D.; Lei, S. Cholecystokinin Facilitates Glutamate Release by Increasing the Number of Readily Releasable Vesicles and Releasing Probability. J. Neurosci. 2010, 30, 5136–5148. [Google Scholar] [CrossRef]
- Lawand, N.B.; Willis, W.D.; Westlund, K.N. Excitatory Amino Acid Receptor Involvement in Peripheral Nociceptive Transmission in Rats. Eur. J. Pharmacol. 1997, 324, 169–177. [Google Scholar] [CrossRef]
- Ferraro, L.; O’connor, W.T.; Li, X.-M.; Rimondini, R.; Beam, L.; Ungerstedt, U.; Fuxe, K.; Tanganelli, S. Evidence for a Differential Cholecystokinin-B and -A Receptor Regulation of Gaba Release in the Rat Nucleus Accumbens Mediated via Dopaminergic and Cholinergic Mechanisms. Neuroscience 1996, 73, 941–950. [Google Scholar] [CrossRef]
- Taylor, B.K.; Joshi, C.; Uppal, H. Stimulation of Dopamine D2 Receptors in the Nucleus Accumbens Inhibits Inflammatory Pain. Brain Res. 2003, 987, 135–143. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y.; Wu, G. Effects of Dopaminergic Agents on Carrageenan Hyperalgesia after Intrathecal Administration to Rats. Eur. J. Pharmacol. 2001, 418, 73–77. [Google Scholar] [CrossRef]
- Barasi, S.; Duggal, K.N. The Effect of Local and Systemic Application of Dopaminergic Agents on Tail Flick Latency in the Rat. Eur. J. Pharmacol. 1985, 117, 287–294. [Google Scholar] [CrossRef]
- Ma, K.-T.; Si, J.-Q.; Zhang, Z.-Q.; Zhao, L.; Fan, P.; Jin, J.-L.; Li, X.-Z.; Zhu, L. Modulatory Effect of CCK-8S on GABA-Induced Depolarization from Rat Dorsal Root Ganglion. Brain Res. 2006, 1121, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Enna, S.J.; McCarson, K.E. The Role of GABA in the Mediation and Perception of Pain. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 54, pp. 1–27. ISBN 978-0-12-032957-1. [Google Scholar]
- Désarmenien, M.; Feltz, P.; Occhipinti, G.; Santangelo, F.; Schlichter, R. Coexistence of GABAA and GABAB Receptors on A Delta and C Primary Afferents. Br. J. Pharm. 1984, 81, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.; Zeilhofer, H.U.; Fritschy, J.-M. Selective Distribution of GABAA Receptor Subtypes in Mouse Spinal Dorsal Horn Neurons and Primary Afferents. J. Comp. Neurol. 2012, 520, 3895–3911. [Google Scholar] [CrossRef]
- Valeyev, A.Y.; Hackman, J.C.; Wood, P.M.; Davidoff, R.A. Pharmacologically Novel GABA Receptor in Human Dorsal Root Ganglion Neurons. J. Neurophysiol. 1996, 76, 3555–3558. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, Y.; Zhao, X.; Chang, Z.; Wei, Y.; Sun, Y.; Jiang, D.; Jiang, X.; Tao, J. Cholecystokinin Type B Receptor-Mediated Inhibition of a-Type K+ Channels Enhances Sensory Neuronal Excitability Through the Phosphatidylinositol 3-Kinase and C-Src-Dependent Jnk Pathway. Cell Commun. Signal. 2019, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemel, B.M.; Ritter, D.M.; Covarrubias, M.; Muqeem, T. A-Type KV Channels in Dorsal Root Ganglion Neurons: Diversity, Function, and Dysfunction. Front. Mol. Neurosci. 2018, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Foucaud, M.; Archer-Lahlou, E.; Marco, E.; Tikhonova, I.G.; Maigret, B.; Escrieut, C.; Langer, I.; Fourmy, D. Insights into the Binding and Activation Sites of the Receptors for Cholecystokinin and Gastrin. Regul. Pept. 2008, 145, 17–23. [Google Scholar] [CrossRef]
- Tikhonova, I.G.; Marco, E.; Lahlou-Archer, E.; Langer, I.; Foucaud, M.; Maigret, B. Daniel Fourmy Validated Ligand Binding Sites in CCK Receptors. Next Step: Computer-Aided Design of Novel CCK Ligands. CTMC 2007, 7, 1243–1247. [Google Scholar] [CrossRef]
- Berna, M.J.; Tapia, J.A.; Sancho, V.; Jensen, R.T. Progress in Developing Cholecystokinin (CCK)/Gastrin Receptor Ligands Which Have Therapeutic Potential. Curr. Opin. Pharm. 2007, 7, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Horwell, D.C.; Hughes, J.; Hunter, J.C.; Pritchard, M.C.; Richardson, R.S.; Roberts, E.; Woodruff, G.N. Rationally Designed “Dipeptoid” Analogues of CCK. Alpha-Methyltryptophan Derivatives as Highly Selective and Orally Active Gastrin and CCK-B Antagonists with Potent Anxiolytic Properties. J. Med. Chem. 1991, 34, 404–414. [Google Scholar] [CrossRef]
- Lotti, V.J.; Chang, R.S. A New Potent and Selective Non-Peptide Gastrin Antagonist and Brain Cholecystokinin Receptor (CCK-B) Ligand: L-365,260. Eur. J. Pharm. 1989, 162, 273–280. [Google Scholar] [CrossRef]
- Hill, D.; Horwell, D.C.; Hunter, J.C.; Kneen, C.O.; Pritchard, M.C.; Suman-Chauhan, N. Synthesis of a Potent and Selective Non-Peptide CCK-B/Gastrin Receptor Antagonist Tritiated Ligand. Bioorganic Med. Chem. Lett. 1993, 3, 885–888. [Google Scholar] [CrossRef]
- Takinami, Y.; Yuki, H.; Nishida, A.; Akuzawa, S.; Uchida, A.; Takemoto, Y.; Ohta, M.; Satoh, M.; Semple, G.; Miyata, K. YF476 Is a New Potent and Selective Gastrin/Cholecystokinin-B Receptor Antagonist In Vitro and In Vivo. Aliment. Pharm. Ther. 1997, 11, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Reeve, J.R.; Liddle, R.A.; McVey, D.C.; Vigna, S.R.; Solomon, T.E.; Keire, D.A.; Rosenquist, G.; Shively, J.E.; Lee, T.D.; Chew, P.; et al. Identification of Nonsulfated Cholecystokinin-58 in Canine Intestinal Extracts and Its Biological Properties. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G326–G333. [Google Scholar] [CrossRef] [Green Version]
- Herranz, R. Cholecystokinin Antagonists: Pharmacological and Therapeutic Potential. Med. Res. Rev. 2003, 23, 559–605. [Google Scholar] [CrossRef] [PubMed]
- Kõks, S.; Fernandes, C.; Kurrikoff, K.; Vasar, E.; Schalkwyk, L.C. Gene Expression Profiling Reveals Upregulation of Tlr4 Receptors in Cckb Receptor Deficient Mice. Behav. Brain Res. 2008, 188, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Kurrikoff, K.; Kõks, S.; Matsui, T.; Bourin, M.; Arend, A.; Aunapuu, M.; Vasar, E. Deletion of the CCK2 Receptor Gene Reduces Mechanical Sensitivity and Abolishes the Development of Hyperalgesia in Mononeuropathic Mice. Eur. J. Neurosci. 2004, 20, 1577–1586. [Google Scholar] [CrossRef]
- Yin, K.; Deuis, J.R.; Lewis, R.J.; Vetter, I. Transcriptomic and Behavioural Characterisation of a Mouse Model of Burn Pain Identify the Cholecystokinin 2 Receptor as an Analgesic Target. Mol. Pain 2016, 12. [Google Scholar] [CrossRef] [Green Version]
- Zubieta, J.-K.; Bueller, J.A.; Jackson, L.R.; Scott, D.J.; Xu, Y.; Koeppe, R.A.; Nichols, T.E.; Stohler, C.S. Placebo Effects Mediated by Endogenous Opioid Activity on μ-Opioid Receptors. J. Neurosci. 2005, 25, 7754–7762. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, F.; Amanzio, M.; Thoen, W. Disruption of Opioid-Induced Placebo Responses by Activation of Cholecystokinin Type-2 Receptors. Psychopharmacology 2011, 213, 791–797. [Google Scholar] [CrossRef]
- Lehmann, K.A.; Schlüsener, M.; Arabatsis, P. Failure of Proglumide, a Cholecystokinin Antagonist, to Potentiate Clinical Morphine Analgesia. A Randomized Double-Blind Postoperative Study Using Patient-Controlled Analgesia (PCA). Anesth. Analg. 1989, 68, 51–56. [Google Scholar]
- Lavigne, G.J.; Hargreaves, K.M.; Schmidt, E.A.; Dionne, R.A. Proglumide Potentiates Morphine Analgesia for Acute Postsurgical Pain. Clin. Pharm. Ther. 1989, 45, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Langley, G.B.; Sheppeard, H. The Visual Analogue Scale: Its Use in Pain Measurement. Rheumatol. Int. 1985, 5, 145–148. [Google Scholar] [CrossRef]
- Bernstein, Z.P.; Yucht, S.; Battista, E.; Lema, M.; Spaulding, M.B. Proglumide as a Morphine Adjunct in Cancer Pain Management. J. Pain Symptom Manag. 1998, 15, 314–320. [Google Scholar] [CrossRef]
- McCleane, G.J. A Randomised, Double Blind, Placebo Controlled Crossover Study of the Cholecystokinin 2 Antagonist L-365,260 as an Adjunct to Strong Opioids in Chronic Human Neuropathic Pain. Neurosci. Lett. 2003, 338, 151–154. [Google Scholar] [CrossRef]
- Hanani, M.; Spray, D.C. Emerging Importance of Satellite Glia in Nervous System Function and Dysfunction. Nat. Rev. Neurosci. 2020, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Cuq, P.; Gross, A.; Terraza, A.; Fourmy, D.; Clerc, P.; Dornand, J.; Magous, R. MRNAs Encding CCKB but Not CCKA Receptors Are Expressed in Human T Lymphocytes and Jurkat Lymphoblastoid Cells. Life Sci. 1997, 61, 543–555. [Google Scholar] [CrossRef]
- Xu, S.-J.; Gao, W.-J.; Cong, B.; Yao, Y.-X.; Gu, Z.-Y. Effect of Lipopolysaccharide on Expression and Characterization of Cholecystokinin Receptors in Rat Pulmonary Interstitial Macrophages. Acta Pharm. Sin. 2004, 25, 1347–1353. [Google Scholar]
- Boyce, M.; Lloyd, K.A.; Pritchard, D.M. Potential Clinical Indications for a CCK2 Receptor Antagonist. Curr. Opin. Pharmacol. 2016, 31, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Novak, D.; Anderluh, M.; Kolenc Peitl, P. CCK2R Antagonists: From SAR to Clinical Trials. Drug Discov. Today 2020, 25, 1322–1336. [Google Scholar] [CrossRef] [PubMed]
- Daugé, V.; Léna, I. CCK in Anxiety and Cognitive Processes. Neurosci. Biobehav. Rev. 1998, 22, 815–825. [Google Scholar] [CrossRef]
- Vialou, V.; Bagot, R.C.; Cahill, M.E.; Ferguson, D.; Robison, A.J.; Dietz, D.M.; Fallon, B.; Mazei-Robison, M.; Ku, S.M.; Harrigan, E.; et al. Prefrontal Cortical Circuit for Depression- and Anxiety-Related Behaviors Mediated by Cholecystokinin: Role of ΔFosB. J. Neurosci. 2014, 34, 3878–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Wang, Y.; Lin, S.; Liu, D.; Mo, G.; Zhang, H.; Dou, Y. Identification of Potential Biomarkers for Abdominal Pain in IBS Patients by Bioinformatics Approach. BMC Gastroenterol. 2021, 21, 48. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Caceres, A.I.; Jordt, S.-E. Activation of a Nerve Injury Transcriptional Signature in Airway-Innervating Sensory Neurons after Lipopolysaccharide-Induced Lung Inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L953–L964. [Google Scholar] [CrossRef]
- Brifault, C.; Romero, H.; Van-Enoo, A.; Pizzo, D.; Azmoon, P.; Kwon, H.; Nasamran, C.; Gonias, S.L.; Campana, W.M. Deletion of the Gene Encoding the NMDA Receptor GluN1 Subunit in Schwann Cells Causes Ultrastructural Changes in Remak Bundles and Hypersensitivity in Pain Processing. J. Neurosci. 2020, 40, 9121–9136. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Agonists | Target | Antagonists | ||||
|---|---|---|---|---|---|---|
| Affinity | Ref | Synthetic | Affinity | Ref | ||
| CCK-4 (endogenous) | Ki = 0.11 µM | [52] | CCK2R | CI-988 (PD-134 308) | Ki = 0.5 nM | [53] |
| L365,260 (benzodiazepine analogue) | Ki = 7 nM | [54] | ||||
| CCK-8 (endogenous) | Ki = 0.3 nM | [14] | Ly225,910 | Ki = 0.2 nM | [55] | |
| Pentagastrin (synthetic) | Ki = 0.0029 µM | [52] | Netazepide (YF476) | Ki = 0.19 nM | [56] | |
| YM-022 | Ki = 68 pM | [52] | ||||
| CCK-8S (endogenous) | CCK1R Ki = 1.41 nM | [57] | CCK1R & CCK2R | Proglumide | CCK1R: IC50 = 6 µM | [58] |
| CCK2R Ki = 0.34 nM | CCK2R: IC50 = 11 µM | |||||
| Model Type | Species | Injury | Treatment | Effect of CCK2R Modulation | Refs |
|---|---|---|---|---|---|
| Central nervous system | |||||
| Traumatic model | Male Sprague–Dawley Rats | Hemisection at T13 on left side | CI-988 (CCK2R antagonist) (i.t.) and systemic injection | CI-988 reduced allodynia | [13] |
| 129S4 CCK2R-/- mice | Chronic constriction injury | No treatment | In midbrain and medulla, CCK2R-/- mice showed reduced expression of MAPK pathway and cytokine production, compared to WT mice | [59] | |
| Peripheral nervous system | |||||
| Traumatic model | 129sv⁄C57BL6 CCK2R-/- mice | Naxolone (opioid receptor antagonist) injection (i.p.) | CCK2R-/- mice showed reduced sensitivity without treatment, naxolone injection increased mechanical allodynia, compared to WT Mice | [60] | |
| L365,260 (i.p.) | High dose of L365,260 decreased the sensitivity of WT mice | [60] | |||
| Ligation of the sciatic nerve | CCK2R-/- mice did not display hyperalgesia, and exhibited less inflammation in sciatic nerve | [60] | |||
| Rats | Unilateral peripheral transection of sciatic nerve | CCK2R was overexpressed in ipsilateral side of axotomy | [28] | ||
| 129sv⁄C57BL6 CCK2R-/- mice | Chronic constriction injury | CCK2R-/- mice had less microglial infiltration in nerve compared to WT | [60] | ||
| Burn model | C57BL6 mice | mild burn injury | Proglumide (30 mg/kg, i.p.), meloxicam (5 mg/kg, i.p.), gabapentin (100 mg/kg, i.p.), oxycodone (10 mg/kg, i.p.) | Proglumide decreased mechanical allodynia alone or with co-administration of oxycodone at low dose (1mg/kg, i.p.) and alleviated thermal allodynia | [61] |
| Diabetic neuropathy model | ICR mice | Diabetic mice (injection of streptozotocin) | CI-988 (i.t.) | CI-988 increased latency of tail response in diabetic mice | [12] |
| CCK-8 (i.t.) | CCK-8 injection decreased latency of tail response in WT mice, reversed by pretreatment with CI-998 | [12] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, A.; Danigo, A.; Bourthoumieu, S.; Mroué, M.; Desmoulière, A.; Sturtz, F.; Rovini, A.; Demiot, C. The Cholecystokinin Type 2 Receptor, a Pharmacological Target for Pain Management. Pharmaceuticals 2021, 14, 1185. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111185
Bernard A, Danigo A, Bourthoumieu S, Mroué M, Desmoulière A, Sturtz F, Rovini A, Demiot C. The Cholecystokinin Type 2 Receptor, a Pharmacological Target for Pain Management. Pharmaceuticals. 2021; 14(11):1185. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111185
Chicago/Turabian StyleBernard, Amandine, Aurore Danigo, Sylvie Bourthoumieu, Mohamad Mroué, Alexis Desmoulière, Franck Sturtz, Amandine Rovini, and Claire Demiot. 2021. "The Cholecystokinin Type 2 Receptor, a Pharmacological Target for Pain Management" Pharmaceuticals 14, no. 11: 1185. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111185