
Advances in Antifungal Drug Development: An Up-To-Date Mini Review



Faculty of Pharmacy in Hradec Králové, Charles University, 50005 Hradec Králové, Czech Republic
*
Authors to whom correspondence should be addressed.
Pharmaceuticals 2021, 14(12), 1312; https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121312
Submission received: 2 December 2021
/
Revised: 11 December 2021
/
Accepted: 14 December 2021
/
Published: 16 December 2021
(This article belongs to the Special Issue Advances in Antifungal Development: Discovery of New Drugs and Drug Repurposing)
Abstract
:The utility of clinically available antifungals is limited by their narrow spectrum of activity, high toxicity, and emerging resistance. Antifungal drug discovery has always been a challenging area, since fungi and their human host are eukaryotes, making it difficult to identify unique targets for antifungals. Novel antifungals in clinical development include first-in-class agents, new structures for an established target, and formulation modifications to marketed antifungals, in addition to repurposed agents. Membrane interacting peptides and aromatherapy are gaining increased attention in the field. Immunotherapy is another promising treatment option, with antifungal antibodies advancing into clinical trials. Novel targets for antifungal therapy are also being discovered, allowing the design of new promising agents that may overcome the resistance issue. In this mini review, we will summarize the current status of antifungal drug pipelines in clinical stages, and the most recent advancements in preclinical antifungal drug development, with special focus on their chemistry.
1. Introduction
For decades, fungal infections have been difficult health conditions to treat. This fact can be attributed to the narrow spectrum and high toxicity of clinically used antifungals, long duration of treatment and the high emergence of resistance towards available agents. The seriousness of fungal infections was brought back to light during the unfortunate COVID-19 pandemic, in the form of secondary life-threatening infections in the intensive care units [1]. Candida, Cryptococcus, and Aspergillus are the most common causative organisms of life-threatening human fungal infections [2]. Candida auris is a multi-drug resistant fungus [3]. Lomentospora prolificans has intrinsic resistance towards all clinically used antifungals. Aspergillus fumigatus is becoming more resistant to treatment, making it more difficult to treat aspergillosis, with mortality rate reaching 100% in some cases. Early detection and treatment of fungal meningitis and chronic pulmonary aspergillosis can save millions of lives around the world. Fungal infections have become a silent crisis, and prompt efforts are needed before it is too late. In this review, we will highlight current state-of-the-art developments in antifungal pipeline, both in clinical and preclinical stages, with special focus on their chemistry, in order to provide the reader with a comprehensive, up-to-date source that will influence future synthetic efforts.
2. Clinically Used Antifungals
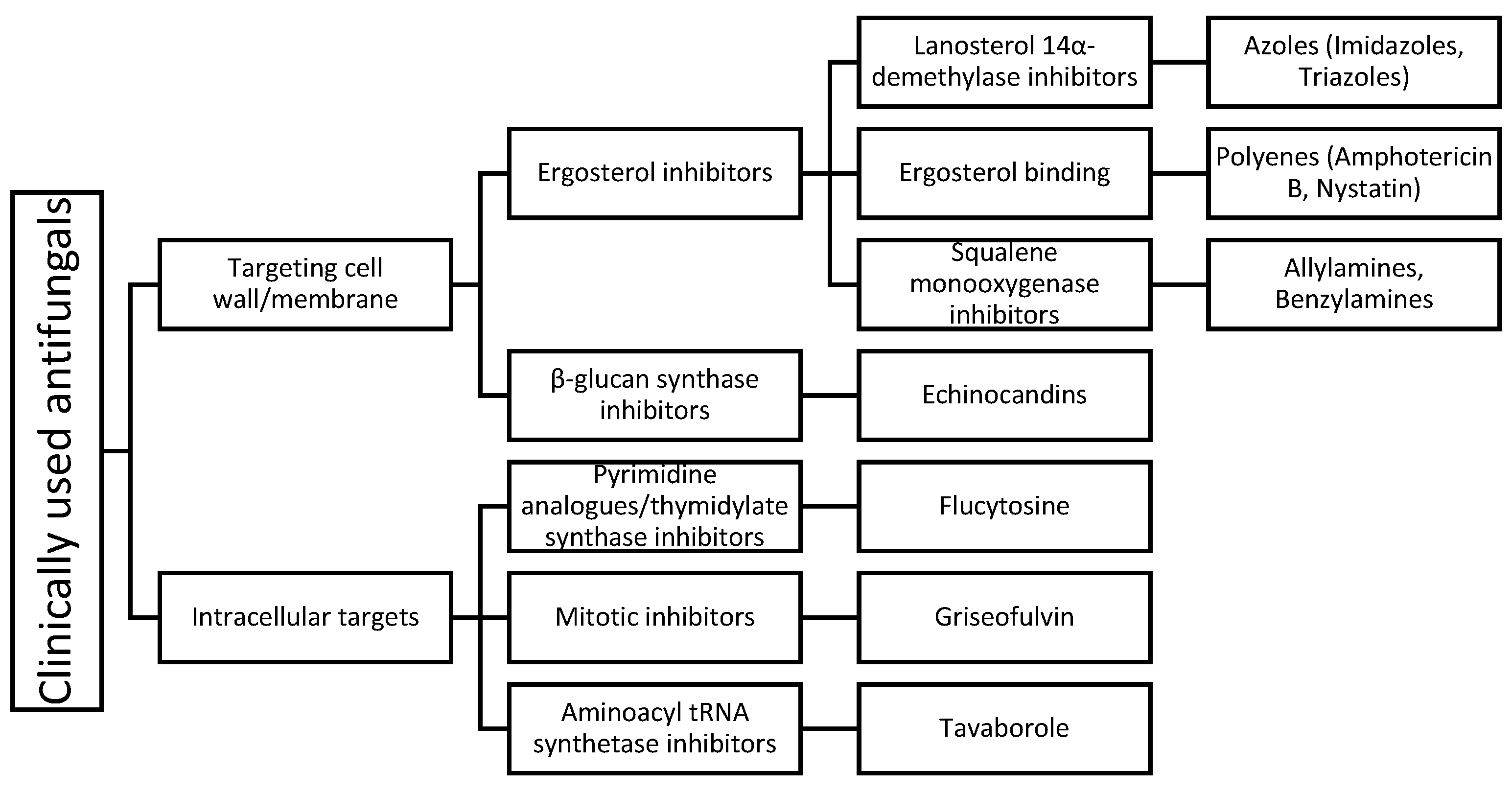
The limited number of available antifungals with there narrow safety margin contributes to the increasing morbidity and mortality of invasive fungal infections. Clinically used antifungals can be classified according to their mechanism of action, as shown in Figure 1.
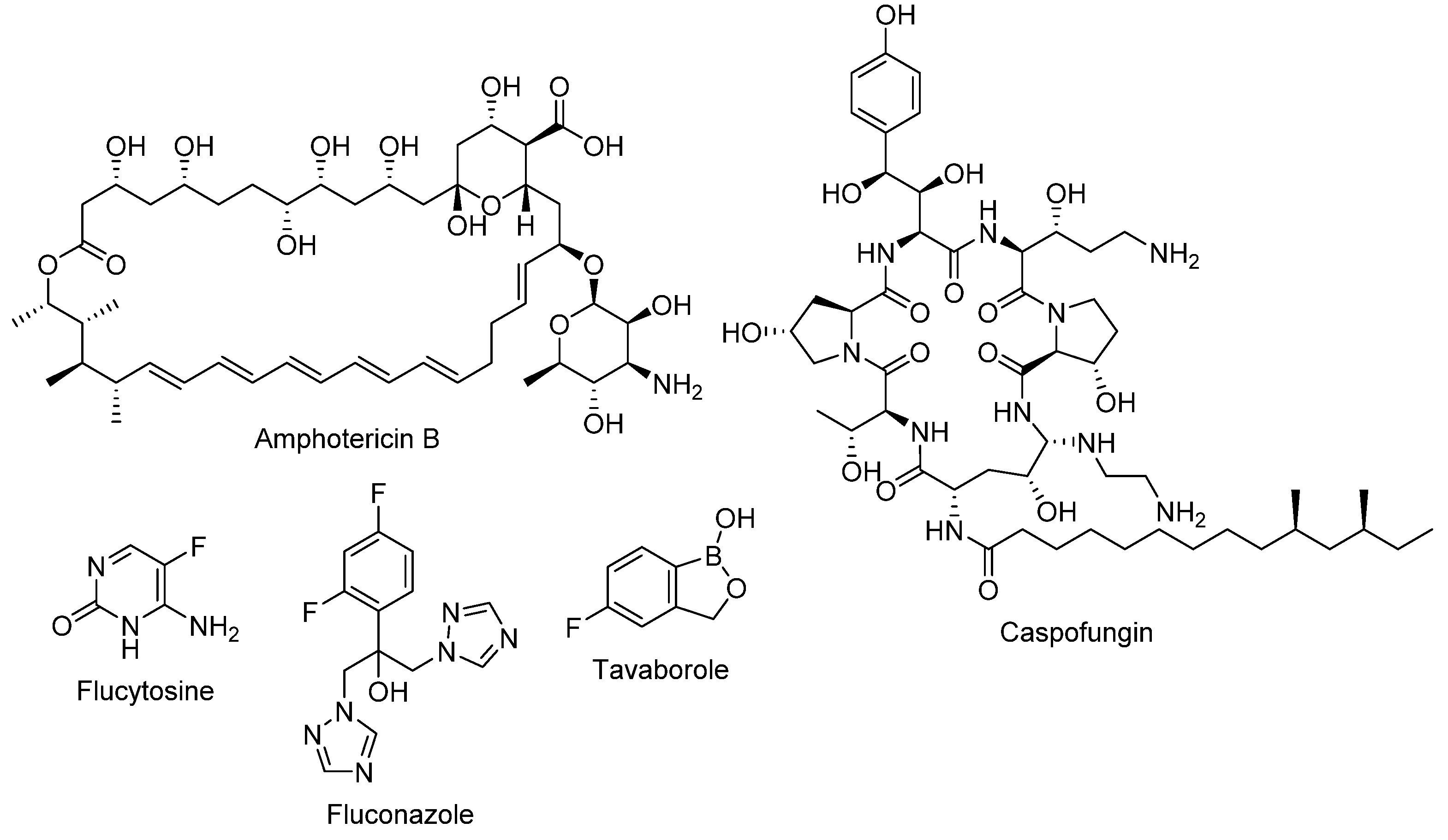
Among all antifungals, only polyenes, flucytosine, azoles, and echinocandins are licensed for the treatment of invasive, life-threatening fungal infections (refer to Figure 2 for chemical structures) [4]. Amphotericin B is the clinically used polyene that is preserved as a second-line agent due to its high nephrotoxicity, which can lead, in some cases, to kidney failure. Several lipid-incorporated formulations of amphotericin B were developed as an attempt to reduce its nephrotoxicity; however, the high cost of such formulations limits their utility. Nystatin is a polyene macrolide antifungal that is used topically or orally to treat oropharyngeal candidiasis (for local effect, as it is not absorbed via the oral route) [5]. The major drawbacks of the pyrimidine antifungal flucytosine are the rapid development of resistance and high toxicity, both hepatological and hematological [6]. Due to their fungistatic mode of action, azoles are associated with a high rate of resistance, yet their wide safety margin contributes to their popularity. Azoles include compounds with either imidazole moiety, such as clotrimazole, or triazole moiety, such as fluconazole. Among the classical antifungals, the echinocandins were the last to be discovered in 1970s, taking them 30 years to progress from bench to bedside, to be marketed later in the year 2000 [7]. Although echinocandins have low rates of resistance, recently some fungi, especially non-albicans candida, started to develop resistance towards echinocandins by acquiring mutations [8]. The long stagnant phase in antifungal development ended with The Food and Drug Administration (FDA) approval of tavaborole in 2014 (refer to Figure 2 for chemical structure). Tavaborole is the first oxaborole and first tRNA synthetase-inhibitor antifungal to be approved for clinical use. It exerts its antifungal activity by inhibiting the cytosolic leucyl-transfer RNA synthetase (LeuRS), which plays a vital role in protein synthesis. It is licensed for topical use for the treatment of onychomycosis [9].
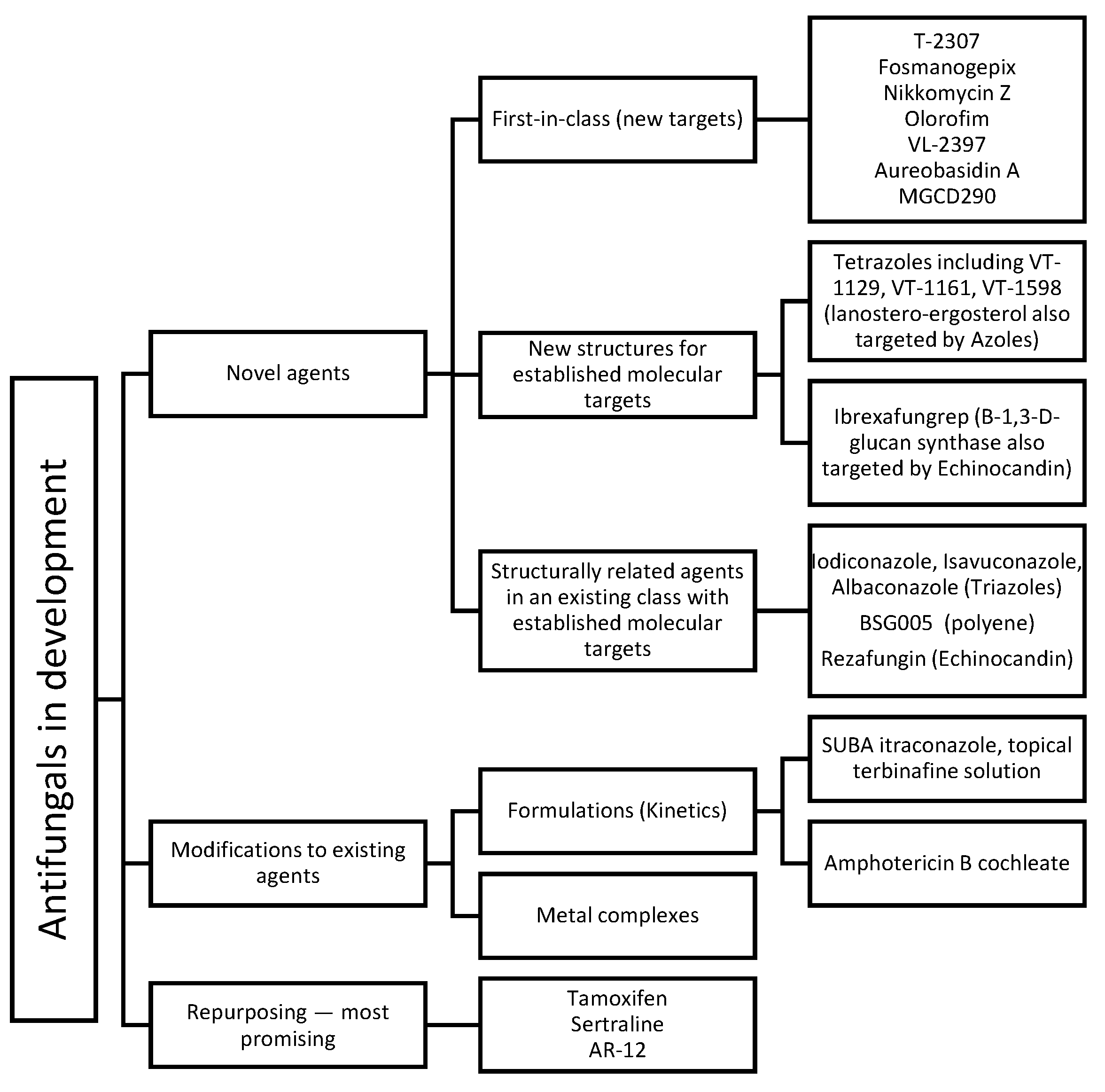
3. Novel Antifungals (In Clinical Settings)
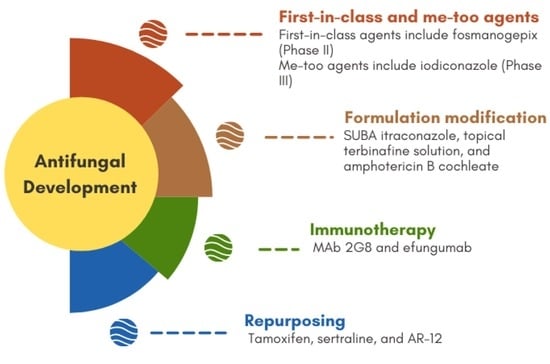
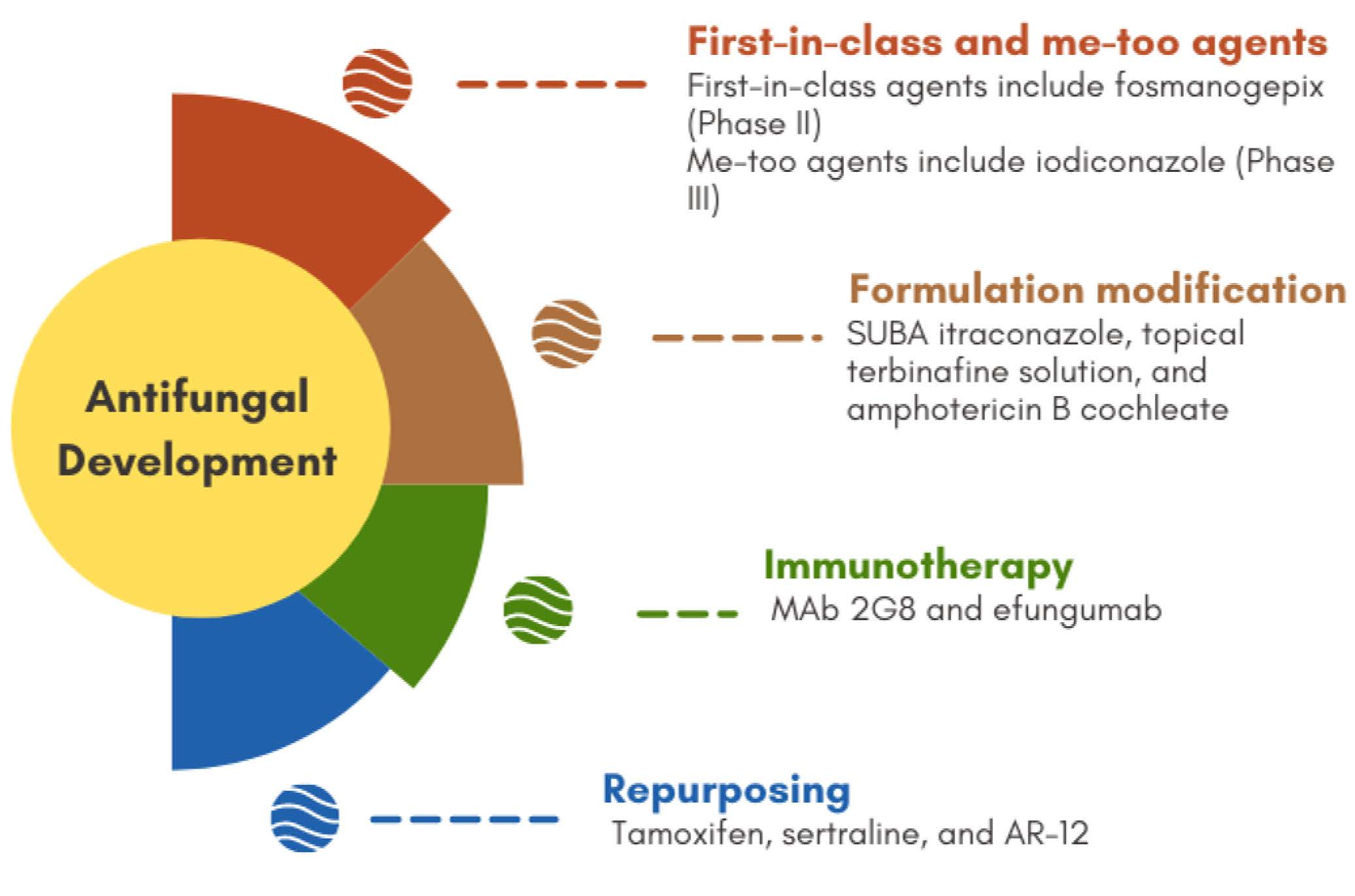
The previously mentioned limitations of available antifungals urge the need for novel agents that can overcome these problems. Novel antifungals include first-in-class agents, new structures for a known target, new derivatives/analogues in an established class of drugs, and modification to the formulations of approved agents, in addition to repurposed agents (refer to Figure 3). Obtaining first-in-class designation significantly speeds up approval process and in-return market availability. Chemistry-wise, new antifungals with new structures include cyclic peptides (hexapeptides such as rezafungin and VL-2397 and depsipeptides such as aureobasidin A), triterpenoids (ibrexafungerp), tetrazoles (VT-1129, VT-1161, VT-1598), orotomides (olorofim), siderophores (VL-2397), and arylamidines (T-2307). Each agent will be discussed separately (ordered according to Figure 3) with a focus on their chemical aspects.
3.1. Aryldiamidines—T-2307
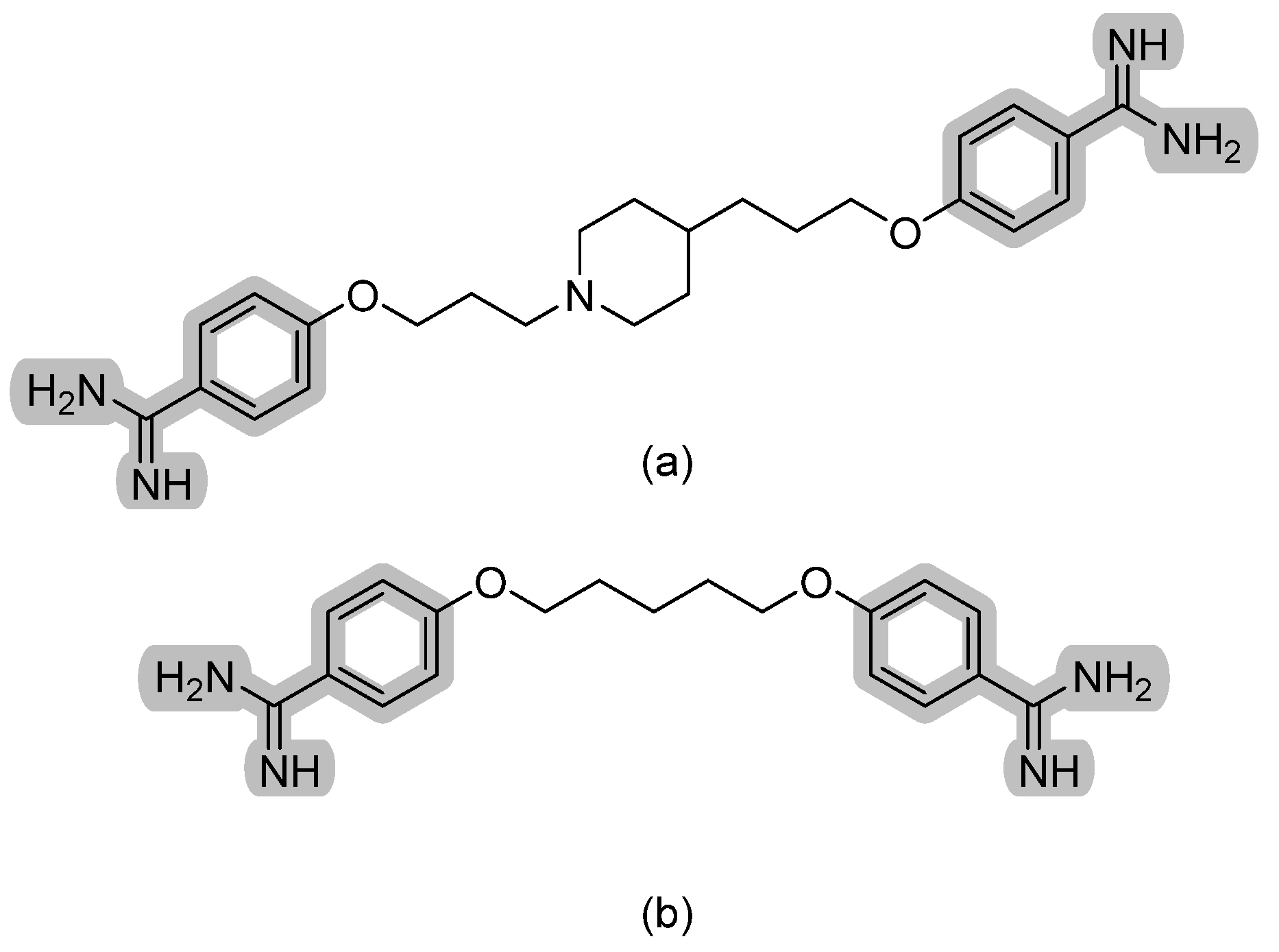
T-2307 is a first-in-class antifungal that exerts its fungicidal activity by inhibiting respiratory chain complexes and thus disrupting mitochondrial membrane potential. It is selectively transported into fungal cells through a polyamine transporter [10]. Structurally, it is an aromatic diamidine that is structurally related to the antiprotozoal agent pentamidine with a characteristic plane of symmetry. Pentamidine is used to treat pneumocystis, leishmaniasis, and trypanosomiasis by a similar mechanism (refer to Figure 4 for chemical structures). Pre-clinical data show T-2307 as very potent antifungal and a perhaps superior agent to azoles and polyenes in the treatment of invasive fungal infections [11]. Further information on T-2307 activity is summarized elsewhere [12].
3.2. Fosmanogepix and Manogepix
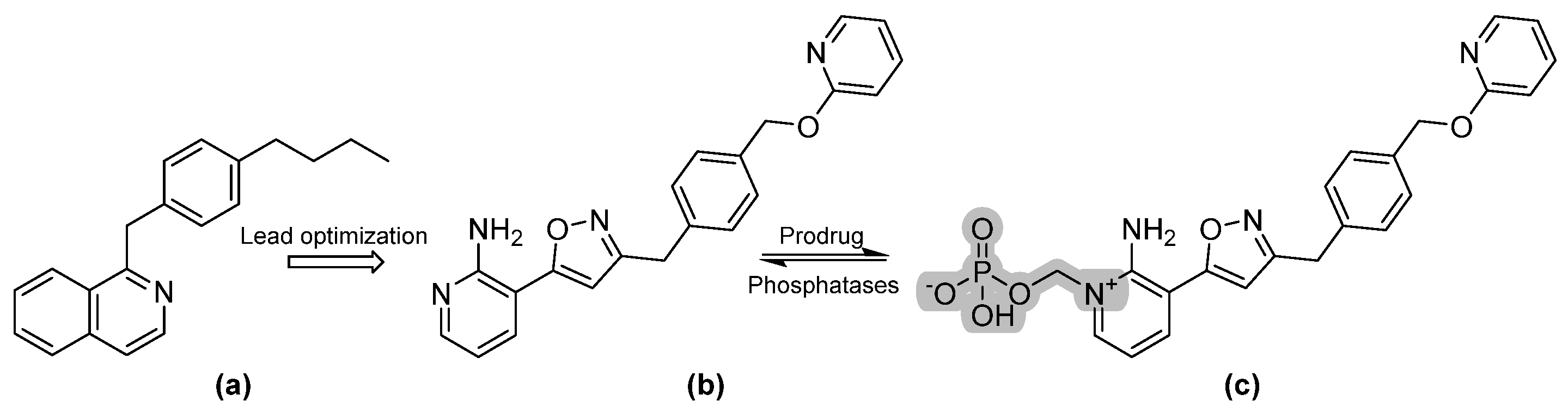
Fosmanogepix is the N-phosphonooxymethylene prodrug of manogepix (APX001A) that is hydrolyzed by systemic phosphatases (refer to Figure 5 for chemical structures). Manogepix was first identified during lead optimization studies to improve the potency of 1-[4-butylbenzyl]isoquinoline, a hit structure found to suppress the expression of surface glycosyl phosphatidylinositol (GPI)-mannoproteins, specifically the Gwt1 protein, in Saccharomyces cerevisiae and Candida albicans, and subsequently inhibit their growth [13,14]. Therefore, manogepix is a first-in-class antifungal that inhibits the fungal Gwt1 protein. Gwt1 is an enzyme that catalyzes inositol acylation, which is an early step in the glycosylphosphatidylinositol (GPI)-anchor biosynthesis pathway [15]. In 2019, fosmanogepix obtained orphan drug designation. Further information on fosmanogepix activity is reviewed elsewhere [16].
3.3. Nikkomycin Z
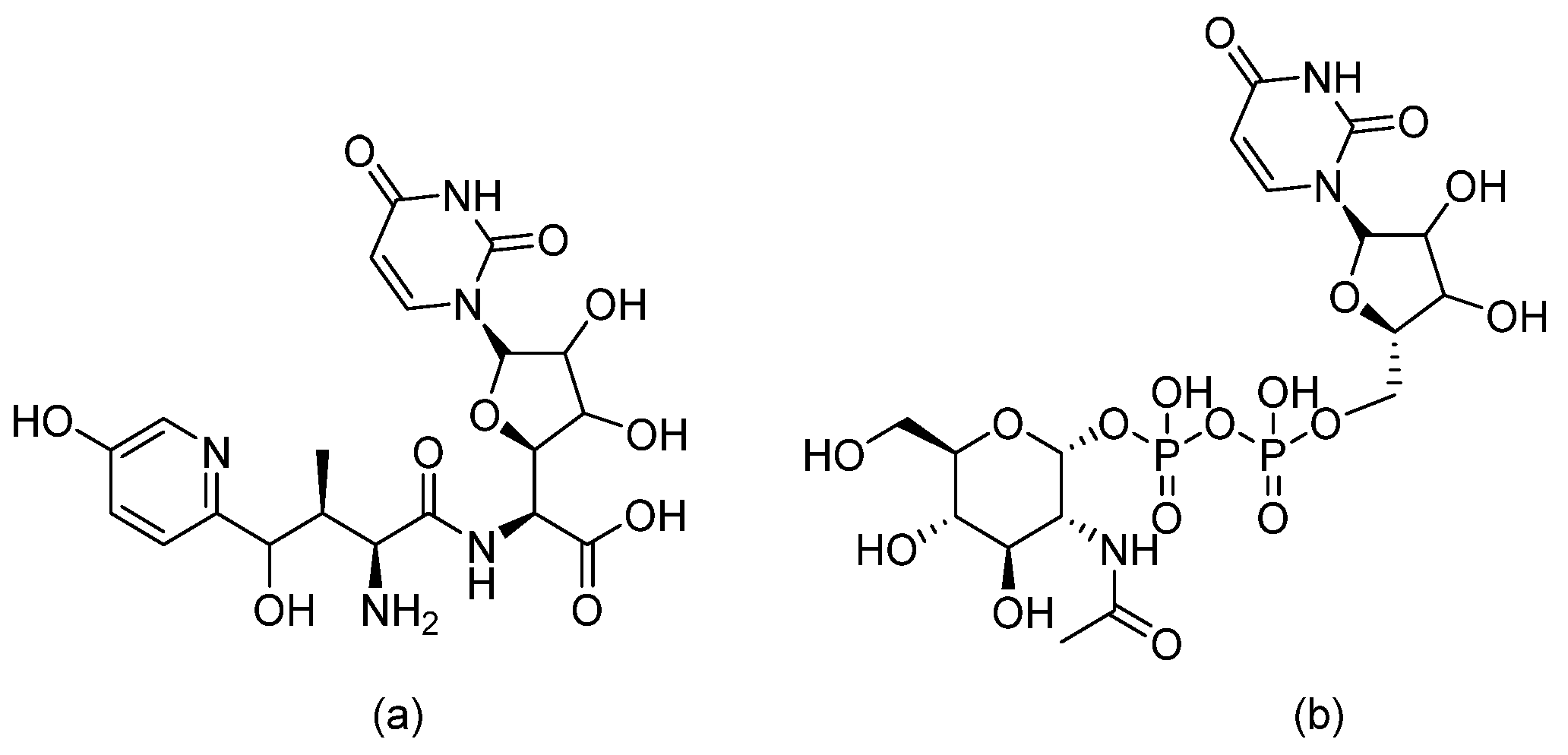
The discovery of nikkomycin Z goes back to the 70s. Structurally, nikkomycin Z resembles uridine diphosphate (UDP)-N-acetyl glucosamine, which is a precursor of chitin, and, thus, nikkomycin Z is a competitive inhibitor of chitin synthase (refer to Figure 6 for chemical structures) [17]. As a stand-alone drug, nikkomycin Z has poor in vitro fungicidal activity against Candida. However, taking into consideration that chitin is a major component of fungal cell walls, nikkomycin Z has synergistic activity with other antifungal cell-wall inhibitors, such as echinocandins [18]. Further information on nikkomycin activity is summarized elsewhere [19].
3.4. Orotomides—Olorofim
The orotomides are a new class of antifungals that exert their fungicidal activity via a novel mechanism of targeting dihydroorotate dehydrogenase, which is a vital enzyme in fungal pyrimidine biosynthesis. Subsequently, nucleic acid and phospholipid synthesis is disrupted [20]. One agent belonging to this new class is olorofim (F901318). Olorofim is a potent antifungal agent that has time-dependent activity against drug resistant Aspergillus spp. and other uncommon molds with promising results in invasive and refractory fungal infections (refer to Figure 7 for chemical structure) [21]. Olorofim is 2000-fold more selective toward fungal dihydroorotate dehydrogenase than the human enzyme homologue [20]. Having a unique mechanism of action and potent antifungal activity against hard-to-treat fungi granted olorifim orphan drug designation (ODD) by the U.S. Food and Drug Administration (FDA) in March 2020 for the treatment of invasive aspergillosis and Lomentospora/Scedosporium infections and later in June 2020 for the treatment of coccidioidomycosis. Similarly, The European Medicines Agency Committee for Orphan Products granted orphan drug status to olorofim for the treatment of invasive aspergillosis and rare mold infections caused by Scedosporium spp. Further information on olorofim’s mechanism of action, pharmacokinetic profile, and clinical efficacy is summarized elsewhere [22].
3.5. Cyclic Peptides—VL-2397
VL-2397 (previously annotated as ASP2397) is a natural cyclic hexapeptide isolated from Acremonium persicinum cultures [23]. It was identified as a potential antifungal while screening various natural secondary metabolites in a silkworm infection model [23]. Structurally, VL-2397 resembles fungal ferrichrome siderophores (high-affinity, iron-chelating compounds secreted from microorganisms that serve as transporters to transport iron across cell membranes) and enters the fungal cells through specific transporters known as siderophore iron transporter 1 (Sit1) (refer to Figure 8 for chemical structures) [24]. Sit1 transporters are not present in mammalian cells, limiting possible toxicity to human cells. Once inside the cell, VL-2397 disrupts important intracellular processes through unknown mechanisms, and hence exerts its fungicidal activity [24].
3.6. Cyclic Peptides—Aureobasidin A
Aureobasidin A is a natural cyclic depsipeptide isolated from the fungus Aureobasidium pullulans R106 that targets the essential inositol phosphorylceramide (sphingolipid) synthase in fungi (refer to Figure 9 for chemical structure) [25]. Interestingly, this broad-spectrum antifungal also exerts significant antiprotozoal activity against the proliferative tachyzoite form of Toxoplasma [26]. Structurally, Aureobasidin A consists of eight α-amino acid units and one hydroxy acid unit. Upon acid hydrolysis, Ikai and colleagues identified these units to be 2(R)-hydroxy-3(R)-methylpentanoic acid, beta-hydroxy-N-methyl-L-valine, N-methyl-L-valine, L-proline, allo-L-isoleucine, N-methyl-L-phenylalanine, L-leucine, and L-phenyl-alanine [25]. Several Aureobasidin A analogues have been prepared by Kurome and colleagues. They found that analogues with 4–6 carbon chain ester derivatives at the γ-carboxyl group of Glu6 or aIle6 exhibited the best antifungal activity [27].
3.7. MGCD290
MGCD290 (chemical structure is not presented in the literature) is an oral fungal Hos2 histone deacetylase (HDAC) inhibitor that also targets non-histone proteins, such as Hsp90, all of which play important roles in gene regulation [28]. Interfering with such fungal proteins suppresses fungal stress responses, which favors the use of MGCD290 as an adjuvant therapy to cell wall/membrane inhibitors [29]. Despite the in vitro synergic activity between MGCD290 and azoles/echinocandins, MGCD290 failed to show clinical importance in clinical settings, and hence its further development was suspended after a phase II clinical trial [28,30].
3.8. Tetrazoles
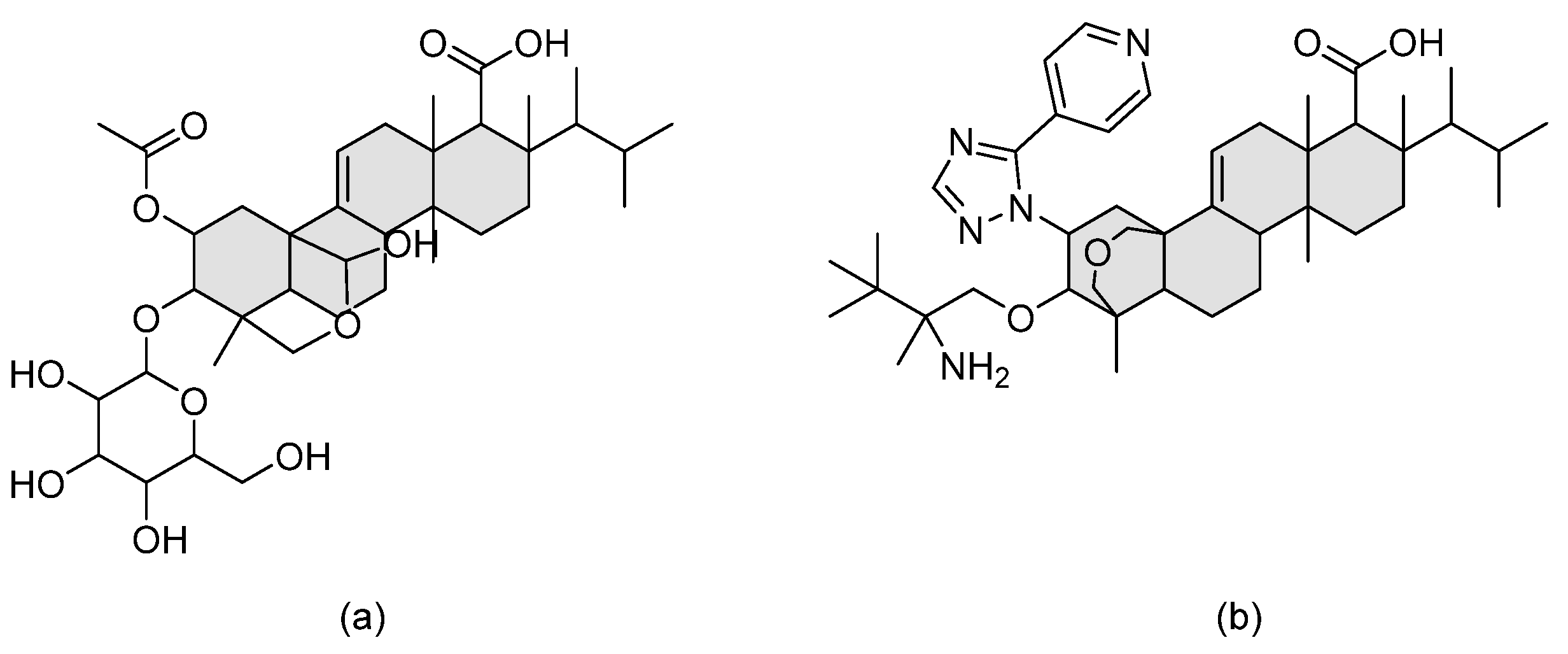
Tetrazoles are a new azole-like group bearing difluoromethyl-pyridines moiety, including the agents VT-1129 (quilseconazole), VT-1161 (oteseconazole), and VT-1598 (refer to Figure 10 for chemical structures). Tetrazoles are more selective to fungal CYP51 (lanosterol demethylase) rather than human enzymes, unlike triazoles, and hence have lower side effects and drug–drug interactions than triazoles [31]. VT-1161 and VT-1598 form H-bonds with the His377 of Candida albicans CYP51 and with the His374 of Aspergillus fumigates CYP51B [32]. The rationale behind the design of tetrazoles by W. J. Hoekstra et al. was to replace the metal binding group in triazoles (MBG, which is triazole) which has strong MBG/metal interaction with another MGB that has weaker MBG/metal interaction (tetrazole), as an attempt to improve selectivity. VT-1129 is a potent oral inhibitor for Cryptococcus species, including drug resistant strains with half-lives of six days [33]. VT-1161 is being investigated as a promising antifungal agent against both fluconazole-sensitive and fluconazole-resistant Candida albicans with clinical application in treating tinea pedis, onychomycosis, and vaginal candidiasis [34]. VT-1161 has half-life of 48 h [34]. VT-1598 was found to have anticandidal activity, especially against Candida auris [35].
3.9. Triterpenoids—Ibrexafungerp
During high-throughput screening of natural products produced by endophytic fungi, enfumafungin was identified as a promising antifungal hemiacetal triterpenoid glycoside. Structural modifications to improve pharmacokinetic properties and, most importantly, oral bioavailability led to the development of the semi-synthetic derivative ibrexafungerp (previously known as SCY-078 or MK-3118) (refer to Figure 11 for chemical structure). Ibrexafungerp exerts its antifungal activity, similarly to echinocandins, via inhibiting β-glucan synthase, yet it has a distinct chemical structure. It is fungicidal against Candida spp. and fungistatic against Aspergillus spp. [36]. Echinocandins are administered intravenously due to their poor per oral stability, which necessitates switching the patient to an oral, and perhaps less potent, antifungal upon discharge. Nevertheless, there is the emerging problem of resistance that urges the need for a safer agent that can be administered at higher doses. Thus, being an orally administered agent, ibrexafungerp is superior to known echinocandins when it comes to patients’ convenience.
3.10. Triazoles
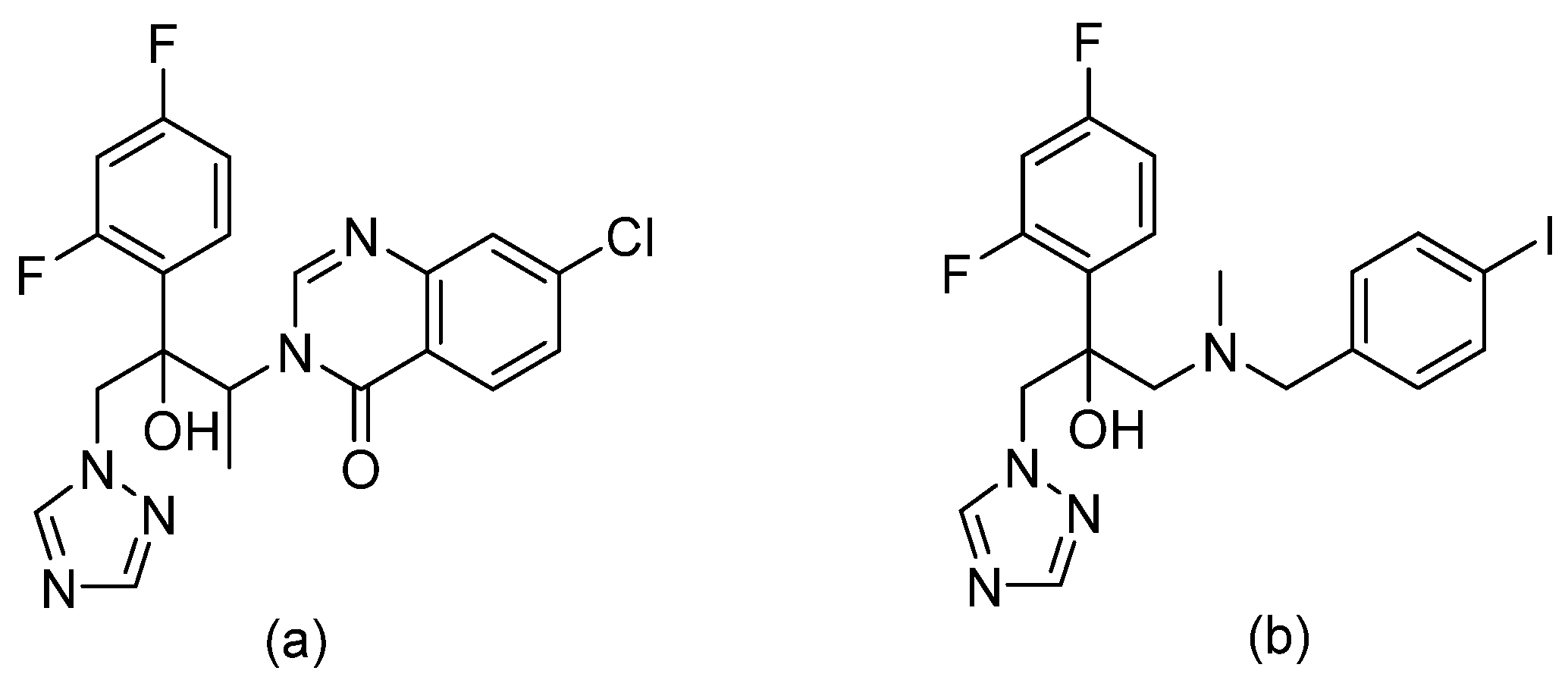
Since they are the most widely used antifungal class, there were several attempts to improve the efficacy of triazoles. Isavuconazonium sulfate is a water-soluble prodrug of isavuconazole, suitable for both oral and IV administration [37]. The [N-(3-acetoxypropyl)-N-methylamino]-carboxymethyl group is linked by an ester functionality to the triazole nitrogen of isavuconazole. The prodrug is cleaved by human plasma esterases releasing isavuconazole and low levels of cleavage by-product (refer to Figure 12) [38]. Isavuconazole has long half-life, broad spectrum of activity, and good efficacy [38]. It was FDA approved in 2015 for the treatment of aspergillosis and invasive mucormycosis.
3.11. BSG005
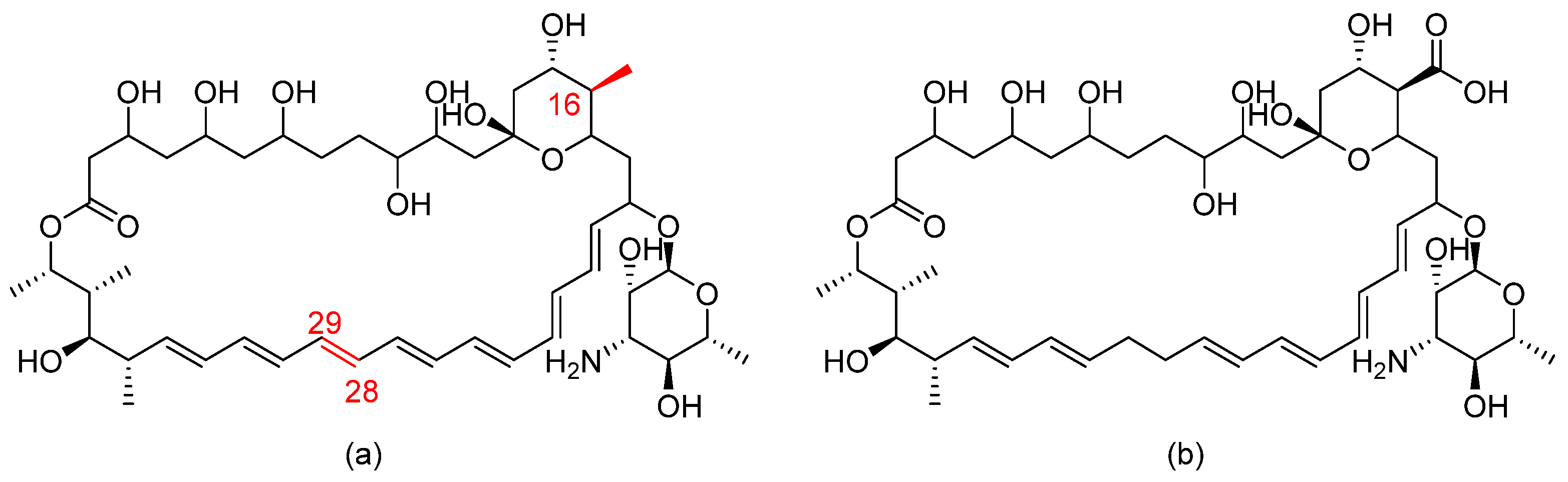
BSG005 is a natural antifungal isolated from Streptomyces noursei. Structurally, BSG005 is an improved version of the polyene nystatin A (heptaene nystatin analogue) (refer to Figure 14 for chemical structures) [41]. It exerts fungicidal activity against a wide range of fungal strains, including azole- and echinocandin- resistant Aspergillus spp. and Candida spp., by a similar mechanism to nystatin. BSG005 does not cause nephrotoxicity, overcoming the main drawback of polyenes [41].
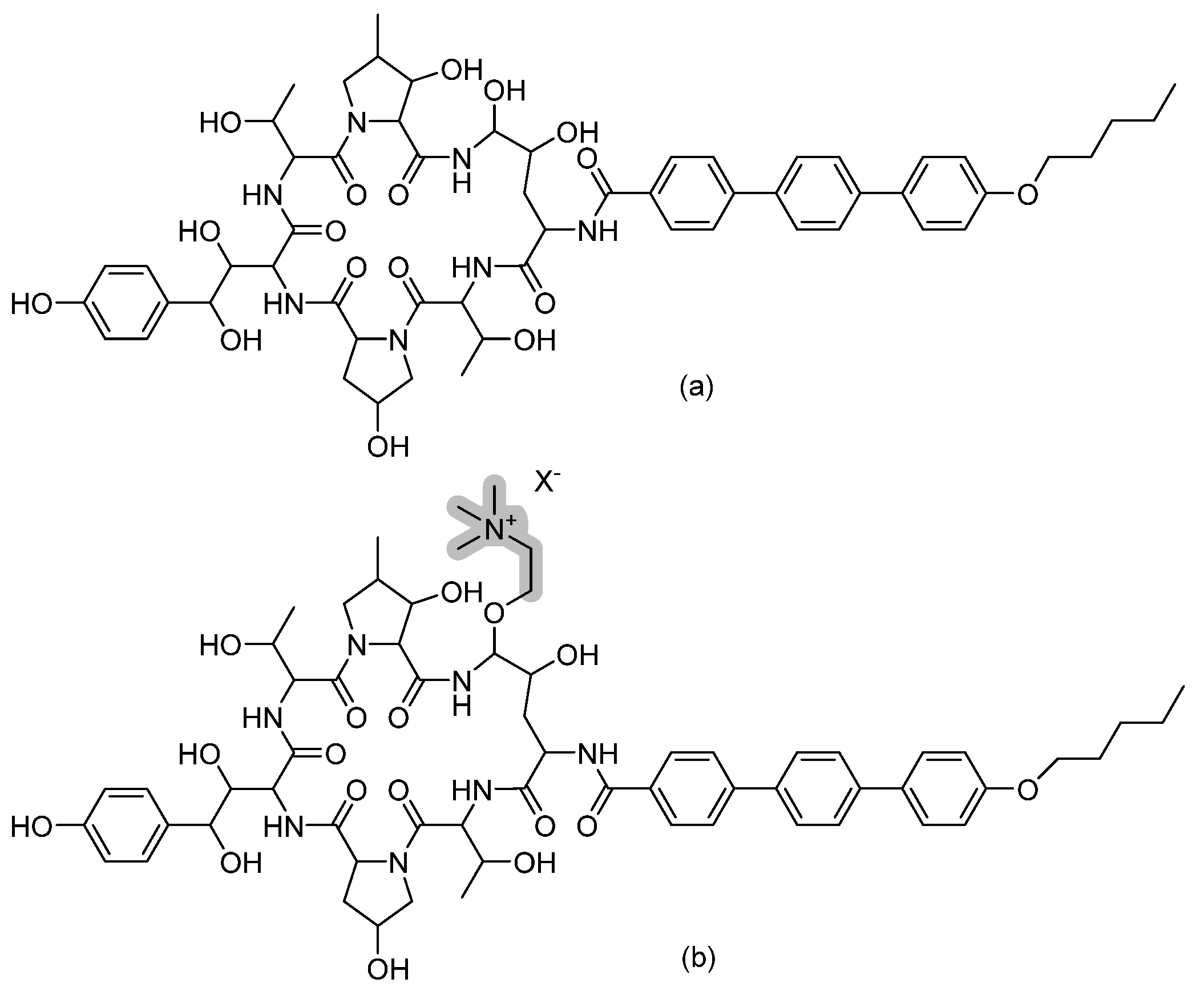
3.12. Cyclic Peptides—Rezafungin
Rezafungin (previously known as CD101) is a novel echinocandin, another cyclic hexapeptide derivative. It is an analogue of the echinocandin anidulafungin obtained by replacing the hemiaminal region at the C5 ornithine position of anidulafungin with choline ether (refer to Figure 15). This chemical modification led to an increased stability towards host degradation and subsequently prolonged the half-life, allowing once-weekly dosing [42]. When incubated in human plasma for 44 h at 37 °C, rezafungin showed a stability of 79–94%, while anidulafungin had 7–15% stability [43]. The prolonged pharmacokinetic property of rezafungin prolonged tissue exposure and hence permitted the use of rezafungin in prophylaxis regimens instead of the regular azoles [44]. This additional choline ether prevented the formation of toxic intermediates, favoring rezafungin safety and allowing its administration at higher doses to prevent resistance [43].
3.13. SUBA Itraconazole
Super-bioavailability-itraconazole (SUBA itraconazole) overcame the low bioavailability issue of itraconazole by dispersing itraconazole in a pH-dependent polymer matrix as capsules. This change to the formulation increased the bioavailability of itraconazole by 173% [45]. SUBA itraconazole was granted FDA approval in 2018 for the treatment of aspergillosis, histoplasmosis, and blastomycosis in patients with contraindications to the use of amphotericin B (intolerant or refractory).
3.14. Topical Terbinafine Solution
Oral terbinafine is a gold-standard treatment for onychomycosis; however the treatment is lengthy, as it takes terbinafine a long time to concentrate in the nail plate and bed, increasing the potential for the development of systemic side effects and resistance. One solution was to prepare terbinafine as a solution for topical use (MOB-01) [46]. Pharmacokinetic studies showed that, when compared with oral terbinafine, topical terbinafine achieves higher concentrations in the nail plate (≈10,003 times more) and nail bed (≈403 times more) with limited or no systemic absorption. Similar examples include two new ophthalmic solutions for the treatment of candida infections; one containing hexamidine diisethionate 0.05% (keratosept) [47], and the other containing povidone-iodine 0.6% (IODIM®) [48].
3.15. Amphotericin B Cochleate
Amphotericin B cochleate is a polyene formulation of amphotericin B that is stable against gastric degradation and hence is suitable for oral administration [49]. Cochleates are made up of phosphatidylserine with phospholipid-calcium precipitates in a multilayer system that has spiral configuration. When the cochleates reach the blood stream, the spiral structure opens once the calcium concentration drop in the cochleate, releasing the encapsulated amphotericin B [49]. The possibility to administer amphotericin B via the oral route overcame infusion-related complications and offered a practical, patient-convenient, broad-spectrum antifungal treatment.
3.16. Metal Complexes and Chelates
Metal complexes may be a novel promising class of antifungals due to improved properties, especially those related to stereochemistry, redox potential, and lipophilicity [50]. Such an approach may provide an easy solution to overcome the issue of fungal resistance. Successful examples of established antifungals complexed with metal ions include organoruthenium complexes conjugated with three different azoles (namely clotrimazole, tioconazole, and miconazole) reported by Kljun et al. [51]. The resulting mono-, bis-, and tris–azoles complexes had millimolar inhibitory concentrations against Culvularia lunata. Another example is reported by Stevanovic et al., who showed that a fluconazole zinc (II) complex had significantly better antifungal activity against Candida krusei and Candida parapsilosis than fluconazole [52]. Other new metal coordinates, not complexed with known antifungals, are at preclinical stages [53,54]. On the other hand, Polvi and colleagues screened a library of pharmacologically active compounds that do not themselves possess antifungal activity as an attempt to identify compounds that can potentiate the efficacy of caspofungin against echinocandin-resistant Candida albicans strains. They identified the broad-spectrum chelator diethylenetriamine pentaacetate (DTPA) as a promising compound that synergizes with capsofungin (refer to Figure 16 for chemical structure). They justified this potentiating activity of DTPA by magnesium chelation [55]. The potential of metal-based compounds as antifungals is thoroughly reviewed elsewhere [56].
The current status for each of the antifungals discussed in this review, including the name of companies and clinical trial registration numbers, are summarized in Table 1 below.
4. New Compounds as Potential Antifungals (In Preclinical Stages)
Several research groups, both in academia and the industry, are focused on developing new, potentially active antifungals. We will briefly mention some of the most recent (published in 2021), successful synthetic efforts from academia (refer to Table 2 for general structures).
Ravu and colleagues recently reported the synthesis and in vitro antifungal evaluation of a series of phloeodictine analogues [59]. Phloeodictines are marine-derived alkaloids, and the phloeodictine-based 6-hydroxy-2,3,4,6-tetrahydropyrrolo[1,2-a]pyrimidinium moiety with an n-tetradecyl side chain at C-6 has shown antifungal activity and serves as a template for further derivatization. They identified three promising analogues (compounds 24, 36, and 48 in the original paper) with potent activity (MIC ≈ 1 µM) against Candida neoformans and low toxicity against mammalian Vero cells (IC50 > 40 μM) [59].
Choi et al. tested their in-house library where they identified a 2-amino-N-(2-(3,4-dichloro-[1,1-biphenyl]-4-yl)ethyl)-pentanamide hydrochloride (compound 22h in the original paper) as a promising hit with potent, fast fungicidal activity against Candida neoformans (MIC ≈ 2.5 µM) and Candida albicans (MIC ≈ 5 µM) [60]. The latter compound also showed synergistic in vitro activity with clinically available antifungals and potent in vivo efficacy in a subcutaneous infection mouse model and an ex vivo human nail infection model. The authors claim that their hit compound exerts its antifungal activity by interfering with fungal cell wall integrity.
On the other hand, Kato et al. prepared thiazoyl guanidine derivatives that inhibit fungal ergosterol biosynthesis [61]. Their hit compound N-(2′-(4-(methylsulfonyl)phenyl)-[4,4′-bithiazol]-2-yl)-tetrahydropyrimidin-2(1H)-imine (compound 6h in original paper) is structurally related to the antifungal abafungin and showed potent in vitro antifungal activity against Aspergillus fumigatus (MIC = 4.7 μM) with a favorable pharmacokinetic profile. In addition, the latter compound exhibited antifungal activity comparable to voriconazole in a murine model of Aspergillus fumigatus infection.
Furthermore, Li and colleagues prepared a series of carboline fungal histone deacetylase (HDAC) inhibitors in an attempt to develop a promising compound for the combinational treatment of azole-resistant candidiasis [62]. Among all synthesized compounds, 2-(4-(3-(8-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-9-yl)propoxy)phenyl)-N-hydroxyacetamide (compound D12 in original paper) showed excellent in vitro and in vivo synergistic antifungal activity with fluconazole to treat azole-resistant candidiasis.
Fungal fatty acid (FA) synthase and desaturase (responsible for introducing double bonds to yield unsaturated FAs) are essential enzymes for the growth and virulence of fungal pathogens [63]. They are structurally distinct from their mammalian homologues, making them suitable targets for antifungal development. DeJarnette et al. performed whole-cell screening using Candida albicans with varying levels of FA synthase or desaturase [64]. They identified four acyl hydrazides as the most promising candidates (compounds 2, 40, 41, and 48 in the original paper) with broad-spectrum activity against Candida albicans, Candida auris, and mucormycetes, including activity against azole-resistant Candida and low in vitro cytotoxicity in HepG2 liver cancer cell line.
Lastly, Lowes and colleagues designed and synthesized dual inhibitors targeting fungal acetohydroxy-acid synthase and NLRP3 inflammasome [65]. Their design rationale was based on the structural similarities between the two types of inhibitors. Fungal acetohydroxy-acid synthase is the common enzyme in the branched-chain amino acid-synthesis pathway [66], while NLRP3 inflammasome is a mammalian cytosolic receptor that mediates innate inflammatory responses to offending fungi by releasing mature interlukein-1 beta (IL-1β) upon activation [67]. The authors established the essential molecular scaffold required for dual activity for the first time, which shall serve as a template for future synthetic efforts. Among their prepared inhibitors, they identified 2-((1-(4-fluorophenyl)-3-oxo-3-phenylpropyl)thio)benzoate (compound 10 in original paper) as the most promising dual inhibitor, since it significantly decreased IL-1β release (IC50 = 2.3 ± 0.8 μM) without affecting mammalian cell viability (viability = 101.5 ± 1.4%) and exerted potent in vitro antifungal activity against Candida albicans (MIC = 6.4 ± 2.6 μM).
Aside from the mentioned small molecules with intracellular targets, membrane-interacting antifungal antimicrobial peptides (AMPs) are another example of a promising class of antifungals. AMPs generally consist of 12 to 54 amino acids with a net positive charge at physiological pH [68]. They are amphipathic in nature, which enhances their interaction with target membranes [69]. Since they target fungal plasma membranes or cell walls, they have the advantage of avoiding intracellular resistance mechanisms. AMPs are selective, with multiple modes of action (for example interaction with membrane phospholipids, sphingolipids, or proteins), and low toxicity to mammalian cells. Detailed information on antifungal AMPs has recently been reviewed elsewhere [70].
In addition, several studies were conducted to explore the role of essential oils in treating fungal infections. In their recent work, Donadu et al. investigated the antifungal activity of the essential oil of the Colombian rue, Ruta graveolens (REO) [71]. They found that REO exerts fungicidal activity against Candida tropicalis and fungistatic activity against Candida albicans by disrupting cellular membrane integrity. REO also showed synergistic activity with amphotericin B [71]. Furthermore, an oil macerate of Helichrysum microphyllum Cambess. subsp. tyrrhenicum Bacch., Brullo & Giusso showed potent inhibiting activity on candida growth, making it a promising agent to be used topically in the treatment of candidiasis [72]. Another example is the essential oil of Austroeupatorium inulaefolium, which showed strong species-dependent antifungal activity against Penicillium brevicompactum and Fusarium oxysporum [73].
5. Repurposing
Repurposing established drugs with possible antifungal activity and pushing them into the antifungal development pipeline saves time and resources, especially given that the pharmacokinetic and pharmacodynamic profiles are already known for such agents. The most promising candidates to be used in antifungal regimens are the selective estrogen receptor modulator tamoxifen (used for breast cancer) and the serotonin reuptake inhibitor sertraline (used for depression) (refer to Figure 17 for chemical structures). Tamoxifen showed anticryptococcal activity and may be a promising synergistic agent with fluconazole [74]. The most advanced repurposing attempt is for sertraline. An ongoing phase III trial is investigating the role of sertraline as an adjuvant therapy to the standard treatment for cryptococcal meningitis (NCT01802385). Furthermore, AR-12 is a celecoxib derivative that was assessed for safety in a phase I oncology clinical trial (refer to Figure 16 for chemical structure). However, it showed consistent antifungal activity against certain yeasts and molds, and thus it was repurposed as a promising adjuvant therapy to fluconazole in the treatment of invasive fungal infections [75].
6. Immunotherapy
Immunotherapy is a new promising strategy to modulate the host immune system and strengthen the innate and adaptive immune response to fight fungal infections. Immunotherapy approaches in treating fungal infections include the administration of recombinant growth factors and cytokines, granulocyte and granulocyte-macrophage colony-stimulating factors, and antibodies. Immunocompromised patients can also benefit from cell therapy, during which innate and adaptive immune cells are introduced to enhance the immune response against the offending fungi. In this review we will focus on antifungal antibodies, which are also known as antifungal passive immunotherapy. Other immunotherapy approaches are reviewed elsewhere [76].
Currently, there only two antifungal antibodies in clinical development. MAb 2G8 is a monoclonal antibody that targets laminarin (consisting mainly of β glucans). It binds to the walls of Candida albicans and Cryptococcus neoformans, inhibiting their growth and capsule formation [77]. Mambro and colleagues reported the development of a new humanized monoclonal antibody derived from MAb 2G8 that specifically targets β-1,3 glucans of pathogenic fungi, such as Candida spp. [78]. The new derivative showed potent in vitro antifungal activity against Candida auris.
Efungumab (also known as mycograb) is a single-chain variable-fragment antibody that targets heat shock protein 90 (HSP90). Efungumab was tested in combination with amphotericin B in clinical settings, where it showed a reduction in the mortality and an improvement in the survival of patients infected with Candida. It must be noted that efungumab was also investigated in clinical trials as an adjuvant to docetaxel in patients with breast cancer (NCT00217815).
One step beyond antifungal antibodies is radio-immunotherapy, where the antifungal antibody is linked to radioisotopes in order to specifically release fungicidal radiation in fungal cells [79]. This approach showed promising results in treating drug-resistant Cryptococcus neoformans infections [79].
7. New Promising Targets for Antifungal Development
Their classification as eukaryotes makes fungal infections a more challenging condition to treat in the drug discovery pipeline. It is important to discover unique targets that are present in fungi and not in humans in order to improve the selectivity and subsequently the safety profile of such agents. One interesting approach to discover new antifungals is what Novartis did by screening their chemical dark matter database for potential antifungals. Chemical dark matter libraries include molecules that have no bioactivity in human targets and thus are of increased value in screening for active molecules against other eukaryotes. During their screening campaign, Novartis have identified a novel antifungal agent and a novel fungal target pathway, which is hemebiosynthesis. The identification of molecular targets opened the window for medicinal chemists to design inhibitors following a target-based drug design approach.
Another example of a new, promising fungal pathway for antifungal drug development is a sphingolipid synthesis pathway. Sphingolipid synthesis is highly preserved among eukaryotes; however, a number of vital structures are unique to fungi, making them promising treatment targets. In fungi, sphingolipids such as inositolphosphoryl ceramide (IPC) and glucosylceramide (GlcCer) play an important role in fungal pathogenicity and fungal growth [80]. Structures that inhibit the enzymes responsible for the synthesis of the latter two sphingolipids were found to disrupt the virulence of Candida albicans, Cryptococcus neoformans, and Aspergillus spp. [81,82]. Promising pre-clinical molecules targeting sphingolipid biosynthesis include sphingosine N-acyltransferase (e.g., australifungin) [83], IPC inhibitors (e.g., the antifungal non-glycosidic macrolide galbonolide A (also known as rustmicin)) [84], GlcCer synthase inhibitors (e.g., D-threo-PDMP) [81], and GlcCer inhibitors (e.g., BHBM) [85], among others (refer to Figure 18 for chemical structures). For such agents, human toxicity is of great concern, except for IPC inhibitors, since mammals do not express IPC [80].
In addition, Krystufek and colleagues described in their latest work the great potential of aspartic proteases, especially Major aspartyl peptidase 1 (May1) secreted from Cryptococcus neoformans, as a potential target for antifungal drug development [86]. Inhibitors of such targets shall be of great benefit, especially in treating cryptococcosis caused by Cryptococcus neoformans or Cryptococcus gattii in immunocompromised patients with HIV, since such inhibitors are also antiretrovirals. In addition to identifying a new promising target, the authors identified promising inhibitors derived from an N-terminally carboxybenzylated phenylstatine scaffold (refer to Figure 18e for general structure). Other fungal targets, such as the Ras pathway, trehalose pathway, metabolic glyoxylate cycle, high-osmolarity glycerol pathway, etc., are also under investigation.
8. Conclusions and Remarks
When comparing older antifungals with the newer ones mentioned in this review, one can sense the influence of the powerful recent advances in structural biology and medicinal chemistry on phenotypic target discovery, drug discovery, and target-based drug design. It must be noted that in order to truly evaluate the efficacy of new antifungals in treating life-threatening infections, infection-specific biomarkers should be relied on rather than death as an endpoint, since such targeted patients have multiple health comorbidities. By doing so, we ensure that no potential hit is lost as a false negative. One important observation from the examples of antifungals in development mentioned earlier in text is the presence of halogen atoms, especially fluorine and chlorine, in their chemical structures. This suggests the significant importance of halogen bonding for the interactions with their corresponding targets.
Funding
This research was funded by EFSA-CDN, grant number CZ.02.1.01/0.0/0.0/16_019/0000841, co-funded by ERDF. The APC was funded by EFSA-CDN, grant number CZ.02.1.01/0.0/0.0/16_019/0000841.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Daria Nawrot for reviewing the manuscript.
Conflicts of Interest
Authors declare no conflicts of interest.
References
- Chowdhary, A.; Tarai, B.; Singh, A.; Sharma, A. Multidrug-Resistant Candida auris Infections in Critically Ill Coronavirus Disease Patients, India, April–July 2020. Emerg. Infect. Dis. 2020, 26, 2694–2696. [Google Scholar] [CrossRef]
- Boral, H.; Metin, B.; Dogen, A.; Seyedmousavi, S.; Ilkit, M. Overview of selected virulence attributes in Aspergillus fumigants, Candida albicans, Cryptococcus neoformans, Trichophyton rubrum, and Exophiala dermatitidis. Fungal Genet. Biol. 2018, 111, 92–107. [Google Scholar] [CrossRef]
- Du, H.; Bing, J.; Hu, T.R.; Ennis, C.L.; Nobile, C.J.; Huang, G.H. Candida auris: Epidemiology, biology, antifungal resistance, and virulence. PLoS Pathog. 2020, 16, e1008921. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Pienaar, E.D.; Young, T.; Holmes, H. Interventions for the prevention and management of oropharyngeal candidiasis associated with HIV infection in adults and children. Cochrane Database Syst. Rev. 2010, 2010, CD003940. [Google Scholar] [CrossRef] [PubMed]
- Vermes, A.; Guchelaar, H.J.; Dankert, J. Flucytosine: A review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 2000, 46, 171–179. [Google Scholar] [CrossRef]
- Emri, T.; Majoros, L.; Toth, V.; Pocsi, I. Echinocandins: Production and applications. Appl. Microbiol. Biotechnol. 2013, 97, 3267–3284. [Google Scholar] [CrossRef]
- Maligie, M.A.; Selitrennikoff, C.P. Cryptococcus neoformans resistance to echinocandins: (1,3)beta-glucan synthase activity is sensitive to echinocandins. Antimicrob. Agents Chemother. 2005, 49, 2851–2856. [Google Scholar] [CrossRef] [Green Version]
- Ciaravino, V.; Coronado, D.; Lanphear, C.; Shaikh, I.; Ruddock, W.; Chanda, S. Tavaborole, a novel boron-containing small molecule for the topical treatment of onychomycosis, is noncarcinogenic in 2-year carcinogenicity studies. Int. J. Toxicol. 2014, 33, 419–427. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakagami, T.; Yamada, E.; Fukuda, Y.; Hayakawa, H.; Nomura, N.; Mitsuyama, J.; Miyazaki, T.; Mukae, H.; Kohno, S. T-2307, a novel arylamidine, is transported into Candida albicans nby a high-affinity spermine and spermidine carrier regulated by Agp2. J. Antimicrob. Chemother. 2016, 71, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Yamada, E.; Nishikawa, H.; Nomura, N.; Mitsuyama, J. T-2307 Shows Efficacy in a Murine Model of Candida glabrata Infection despite In Vitro Trailing Growth Phenomena. Antimicrob. Agents Chemother. 2010, 54, 3630–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederhold, N.P. Review of T-2307, an Investigational Agent That Causes Collapse of Fungal Mitochondrial Membrane Potential. J. Fungi 2021, 7, 130. [Google Scholar] [CrossRef]
- Mann, P.A.; McLellan, C.A.; Koseoglu, S.; Si, Q.; Kuzmin, E.; Flattery, A.; Harris, G.; Sher, X.; Murgolo, N.; Wang, H.; et al. Chemical Genomics-Based Antifungal Drug Discovery: Targeting Glycosylphosphatidylinositol (GPI) Precursor Biosynthesis. ACS Infect. Dis. 2015, 1, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Tsukada, I.; Tanaka, K.; Matsukura, M.; Haneda, T.; Inoue, S.; Murai, N.; Abe, S.; Ueda, N.; Miyazaki, M.; et al. Synthesis and evaluation of novel antifungal agents-quinoline and pyridine amide derivatives. Bioorganic Med. Chem. Lett. 2010, 20, 4624–4626. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.A.; Miyazaki, M.; Horii, T.; Sagane, K.; Tsukahara, K.; Hata, K. E1210, a new broad-spectrum antifungal, suppresses Candida albicans hyphal growth through inhibition of glycosylphosphatidylinositol biosynthesis. Antimicrob. Agents Chemother. 2011, 56, 960–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, K.J.; Ibrahim, A.S. Fosmanogepix: A Review of the First-in-Class Broad Spectrum Agent for the Treatment of Invasive Fungal Infections. J. Fungi 2020, 6, 239. [Google Scholar] [CrossRef] [PubMed]
- Hector, R.F.; Pappagianis, D. Inhibition of Chitin Synthesis in the Cell-Wall of Coccidioides-Immitis by Polyoxin-D. J. Bacteriol. 1983, 154, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Fortwendel, J.R.; Juvvadi, P.R.; Pinchai, N.; Perfect, B.Z.; Alspaugh, J.A.; Perfect, J.R.; Steinbach, W.J. Differential Effects of Inhibiting Chitin and 1,3-beta-D-Glucan Synthesis in Ras and Calcineurin Mutants of Aspergillus fumigatus. Antimicrob. Agents Chemother. 2009, 53, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Larwood, D.J. Nikkomycin Z-Ready to Meet the Promise? J. Fungi 2020, 6, 261. [Google Scholar] [CrossRef]
- Oliver, J.D.; Sibley, G.; Beckmann, N.; Dobb, K.S.; Slater, M.J.; McEntee, L.; du Pré, S.; Livermore, J.; Bromley, M.J.; Wiederhold, N.P.; et al. F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc. Natl. Acad. Sci. USA 2016, 113, 12809–12814. [Google Scholar] [CrossRef] [Green Version]
- Negri, C.E.; Johnson, A.; McEntee, L.; Box, H.; Whalley, S.; Schwartz, J.A.; Ramos-Martín, V.; Livermore, J.; Kolamunnage-Dona, R.; Colombo, A.L.; et al. Pharmacodynamics of the Novel Antifungal Agent F901318 for Acute Sinopulmonary Aspergillosis Caused by Aspergillus flavus. J. Infect. Dis. 2018, 217, 1118–1127. [Google Scholar] [CrossRef]
- Wiederhold, N.P. Review of the Novel Investigational Antifungal Olorofim. J. Fungi 2020, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Kanasaki, R.; Yoshikawa, K.; Furukawa, S.; Fujie, A.; Hamamoto, H.; Sekimizu, K. Discovery of a new antifungal agent ASP2397 using a silkworm model of Aspergillus fumigatus infection. J. Antibiot. (Tokyo) 2017, 70, 41–44. [Google Scholar] [CrossRef]
- Nakamura, I.; Ohsumi, K.; Takeda, S.; Katsumata, K.; Matsumoto, S.; Akamatsu, S.; Mitori, H.; Nakai, T. ASP2397 Is a Novel Natural Compound That Exhibits Rapid and Potent Fungicidal Activity against Aspergillus Species through a Specific Transporter. Antimicrob. Agents Chemother. 2019, 63, e02689-18. [Google Scholar] [CrossRef]
- Ikai, K.; Takesako, K.; Shiomi, K.; Moriguchi, M.; Umeda, Y.; Yamamoto, J.; Kato, I.; Naganawa, H. Structure of aureobasidin A. J. Antibiot. 1991, 44, 925–933. [Google Scholar] [CrossRef]
- Alqaisi, A.; Mbekeani, A.J.; Llorens, M.B.; Elhammer, A.P.; Denny, P.W. The antifungal Aureobasidin A and an analogue are active against the protozoan parasite Toxoplasma gondii but do not inhibit sphingolipid biosynthesis—Corrigendum. Parasitology 2018, 145, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurome, T.; Inoue, T.; Takesako, K.; Kato, I.; Inami, K.; Shiba, T. Syntheses of antifungal aureobasidin A analogs with alkyl chains for structure-activity relationship. J. Antibiot. 1998, 51, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, M.A.; Messer, S.A.; Georgopapadakou, N.; Martell, L.A.; Besterman, J.M.; Diekema, D.J. Activity of MGCD290, a Hos2 Histone Deacetylase Inhibitor, in Combination with Azole Antifungals against Opportunistic Fungal Pathogens. J. Clin. Microbiol. 2009, 47, 3797–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowen, L.E. The fungal Achilles’ heel: Targeting Hsp90 to cripple fungal pathogens. Curr. Opin. Microbiol. 2013, 16, 377–384. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Rhomberg, P.R.; Messer, S.A.; Castanheira, M. In vitro activity of a Hos2 deacetylase inhibitor, MGCD290, in combination with echinocandins against echinocandin-resistant Candida species. Diagn. Microbiol. Infect. Dis. 2015, 81, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef]
- Zhang, J.X.; Li, L.P.; Lv, Q.Z.; Yan, L.; Wang, Y.; Jiang, Y.Y. The Fungal CYP51s: Their Functions, Structures, Related Drug Resistance, and Inhibitors. Front. Microbiol. 2019, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The Investigational Drug VT-1129 Is a Highly Potent Inhibitor of Cryptococcus Species CYP51 but Only Weakly Inhibits the Human Enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef] [Green Version]
- Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Sobel, J.D.; Lilly, E.A.; Fidel, P.L., Jr. Efficacy of the Clinical Agent VT-1161 against Fluconazole-Sensitive and -Resistant Candida albicans in a Murine Model of Vaginal Candidiasis. Antimicrob. Agents Chemother. 2015, 59, 5567–5573. [Google Scholar] [CrossRef] [Green Version]
- Wiederhold, N.P.; Lockhart, S.R.; Najvar, L.K.; Berkow, E.L.; Jaramillo, R.; Olivo, M.; Garvey, E.P.; Yates, C.M.; Schotzinger, R.J.; Catano, G.; et al. The Fungal Cyp51-Specific Inhibitor VT-1598 Demonstrates In Vitro and In Vivo Activity against Candida auris. Antimicrob. Agents Chemother. 2019, 63, e02233-18. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.C.; Hicks, P.S.; Kurtz, M.B.; Rosen, H.; Schmatz, D.M.; Liberator, P.A.; Douglas, C.M. The antifungal echinocandin caspofungin acetate kills growing cells of Aspergillus fumigatus in vitro. Antimicrob. Agents Chemother. 2002, 46, 3001–3012. [Google Scholar] [CrossRef] [Green Version]
- Odds, F.C. Drug evaluation: BAL-8557—A novel broad-spectrum triazole antifungal. Curr. Opin. Investig. Drugs 2006, 7, 766–772. [Google Scholar] [PubMed]
- Schmitt-Hoffmann, A.; Roos, B.; Heep, M.; Schleimer, M.; Weidekamm, E.; Brown, T.; Roehrle, M.; Beglinger, C. Single-ascending-dose pharmacokinetics and safety of the novel broad-spectrum antifungal triazole BAL4815 after intravenous infusions (50, 100, and 200 milligrams) and oral administrations (100, 200, and 400 milligrams) of its prodrug, BAL8557, in healthy volunteers. Antimicrob. Agents Chemother. 2006, 50, 279–285. [Google Scholar]
- Bartroli, J.; Turmo, E.; Algueró, M.; Boncompte, E.; Vericat, M.L.; Conte, L.; Ramis, J.; Merlos, M.; García-Rafanell, J.; Forn, J. New azole antifungals. 2. Synthesis and antifungal activity of heterocyclecarboxamide derivatives of 3-amino-2-aryl-1-azolyl-2-butanol. J. Med. Chem. 1998, 41, 1855–1868. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Xie, Y.; Sheng, C.; Cao, Y.; Zhang, W.; Chen, H.; Fan, G. In vivo pharmacokinetics and in vitro antifungal activity of iodiconazole, a new triazole, determined by microdialysis sampling. Int. J. Antimicrob. Agents 2013, 41, 229–235. [Google Scholar] [CrossRef]
- Brautaset, T.; Sletta, H.; Degnes, K.F.; Sekurova, O.N.; Bakke, I.; Volokhan, O.; Andreassen, T.; Ellingsen, T.E.; Zotchev, S.B. New nystatin-related antifungal polyene macrolides with altered polyol region generated via biosynthetic engineering of Streptomyces noursei. Appl. Environ. Microbiol. 2011, 77, 6636–6643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, B.R.; James, K.D.; Polowy, K.; Bryant, B.J.; Vaidya, A.; Smith, S.; Laudeman, C.P. CD101, a novel echinocandin with exceptional stability properties and enhanced aqueous solubility. J. Antibiot. 2017, 70, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Ong, V.; James, K.D.; Smith, S.; Krishnan, B.R. Pharmacokinetics of the Novel Echinocandin CD101 in Multiple Animal Species. Antimicrob. Agents Chemother. 2017, 61, e01626-16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Prideaux, B.; Nagasaki, Y.; Lee, M.H.; Chen, P.Y.; Blanc, L.; Ho, H.; Clancy, C.J.; Nguyen, M.H.; Dartois, V.; et al. Unraveling Drug Penetration of Echinocandin Antifungals at the Site of Infection in an Intra-abdominal Abscess Model. Antimicrob. Agents Chemother. 2017, 61, e01009-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuhelwa, A.Y.; Foster, D.J.R.; Mudge, S.; Hayes, D.; Upton, R.N. Population Pharmacokinetic Modeling of Itraconazole and Hydroxyitraconazole for Oral SUBA-Itraconazole and Sporanox Capsule Formulations in Healthy Subjects in Fed and Fasted States. Antimicrob. Agents Chemother. 2015, 59, 5681–5696. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Surprenant, M.S.; Kempers, S.E.; Pariser, D.M.; Rensfeldt, K.; Tavakkol, A. Efficacy and safety of topical terbinafine 10% solution (MOB-015) in the treatment of mild to moderate distal subungual onychomycosis: A randomized, multicenter, double-blind, vehicle-controlled phase 3 study. J. Am. Acad. Dermatol. 2021, 85, 95–104. [Google Scholar] [CrossRef]
- Pinna, A.; Donadu, M.G.; Usai, D.; Dore, S.; Boscia, F.; Zanetti, S. In Vitro Antimicrobial Activity of a New Ophthalmic Solution Containing Hexamidine Diisethionate 0.05% (Keratosept). Cornea 2020, 39, 1415–1418. [Google Scholar] [CrossRef]
- Pinna, A.; Donadu, M.G.; Usai, D.; Dore, S.; D’Amico-Ricci, G.; Boscia, F.; Zanetti, S. In vitro antimicrobial activity of a new ophthalmic solution containing povidone-iodine 0.6% (IODIM (R)). Acta Ophthalmol. 2020, 98, E178–E180. [Google Scholar] [CrossRef]
- Santangelo, R.; Paderu, P.; Delmas, G.; Chen, Z.W.; Mannino, R.; Zarif, L.; Perlin, D.S. Efficacy of oral cochleate-amphotericin B in a mouse model of systemic candidiasis. Antimicrob. Agents Chemother. 2000, 44, 2356–2360. [Google Scholar] [CrossRef] [Green Version]
- Gasser, G.; Metzler-Nolte, N. The potential of organometallic complexes in medicinal chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Kljun, J.; Scott, A.J.; Rizner, T.L.; Keiser, J.; Turel, I. Synthesis and Biological Evaluation of Organoruthenium Complexes with Azole Antifungal Agents. First Crystal Structure of a Tioconazole Metal Complex. Organometallics 2014, 33, 1594–1601. [Google Scholar] [CrossRef]
- Stevanović, N.L.; Aleksic, I.; Kljun, J.; Bogojevic, S.S.; Veselinovic, A.; Nikodinovic-Runic, J.; Turel, I.; Djuran, M.I.; Glišić, B.Đ. Copper(II) and Zinc(II) Complexes with the Clinically Used Fluconazole: Comparison of Antifungal Activity and Therapeutic Potential. Pharmaceuticals 2021, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Andrejević, T.P.; Aleksic, I.; Počkaj, M.; Kljun, J.; Milivojevic, D.; Stevanović, N.L.; Nikodinovic-Runic, J.; Turel, I.; Djuran, M.I.; Glišić, B.Đ. Tailoring copper(II) complexes with pyridine-4,5-dicarboxylate esters for anti-Candida activity. Dalton Trans. 2021, 50, 2627–2638. [Google Scholar] [CrossRef]
- Savic, N.D.; Vojnovic, S.; Glisic, B.Ð.; Crochet, A.; Pavic, A.; Janjic, G.V.; Pekmezovic, M.; Opsenica, I.M.; Fromm, K.M.; Nikodinovic-Runic, J.; et al. Mononuclear silver(I) complexes with 1,7-phenanthroline as potent inhibitors of Candida growth. Eur. J. Med. Chem. 2018, 156, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Polvi, E.J.; Averette, A.F.; Lee, S.C.; Kim, T.; Bahn, Y.; Veri, A.O.; Robbins, N.; Heitman, J.; Cowen, L.E. Metal Chelation as a Powerful Strategy to Probe Cellular Circuitry Governing Fungal Drug Resistance and Morphogenesis. PLoS Genet. 2016, 12, e1006350. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Betts, H.; Keller, S.; Cariou, K.; Gasser, G. Recent developments of metal-based compounds against fungal pathogens. Chem. Soc. Rev. 2021, 50, 10346–10402. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Fukuda, Y.; Mitsuyama, J.; Tashiro, M.; Tanaka, A.; Takazono, T.; Saijo, T.; Yamamoto, K.; Nakamura, S.; Imamura, Y.; et al. In vitro and in vivo antifungal activities of T-2307, a novel arylamidine, against Cryptococcus gattii: An emerging fungal pathogen. J. Antimicrob. Chemother. 2017, 72, 1709–1713. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Wen, J.; Lu, G.; Hong, Z.; Fan, G.; Wu, Y.; Sheng, C.; Zhang, W. An ultra-fast LC method for the determination of iodiconazole in microdialysis samples and its application in the calibration of laboratory-made linear probes. J. Pharm. Biomed. Anal. 2010, 51, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Ravu, R.R.; Jacob, M.R.; Khan, S.I.; Wang, M.; Cao, L.; Agarwal, A.K.; Clark, A.M.; Li, X. Synthesis and Antifungal Activity Evaluation of Phloeodictine Analogues. J. Nat. Prod. 2021, 84, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Lee, K.; Kim, S.; Lee, Y.R.; Kim, H.J.; Seo, K.J.; Lee, M.H.; Yeon, S.K.; Jang, B.K.; Park, S.J.; et al. Optimization and Evaluation of Novel Antifungal Agents for the Treatment of Fungal Infection. J. Med. Chem. 2021, 64, 15912–15935. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Ukai, Y.; Kondo, N.; Nozu, K.; Kimura, C.; Hashimoto, K.; Mizusawa, E.; Maki, H.; Naito, A.; Kawai, M. Identification of Thiazoyl Guanidine Derivatives as Novel Antifungal Agents Inhibiting Ergosterol Biosynthesis for Treatment of Invasive Fungal Infections. J. Med. Chem. 2021, 64, 10482–10496. [Google Scholar]
- Li, Z.; Tu, J.; Han, G.Y.; Liu, N.; Sheng, C.Q. Novel Carboline Fungal Histone Deacetylase (HDAC) Inhibitors for Combinational Treatment of Azole-Resistant Candidiasis. J. Med. Chem. 2021, 64, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Plaine, A.; Albert, J.; Prasad, T.; Prasad, R.; Ernst, J.F. Dosage-dependent functions of fatty acid desaturase Ole1p in growth and morphogenesis of Candida albicans. Microbiology-Sgm 2004, 150, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- DeJarnette, C.; Meyer, C.J.; Jenner, A.R.; Butts, A.; Peters, T.; Cheramie, M.N.; Phelps, G.A.; Vita, N.A.; Loudon-Hossler, V.C.; Lee, R.E.; et al. Identification of Inhibitors of Fungal Fatty Acid Biosynthesis. ACS Infect. Dis. 2021, 7, 3210–3223. [Google Scholar] [CrossRef] [PubMed]
- Lowes, D.J.; Miao, J.; Al-Waqfi, R.A.; Avad, K.A.; Hevener, K.E.; Peters, B.M. Identification of Dual-Target Compounds with Anti-fungal and Anti-NLRP3 Inflammasome Activity. ACS Infect. Dis. 2021, 7, 2522–2535. [Google Scholar] [CrossRef] [PubMed]
- Pue, N.; Guddat, L.W. Acetohydroxyacid Synthase: A Target for Antimicrobial Drug Discovery. Curr. Pharm. Des. 2014, 20, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Camilli, G.; Griffiths, J.S.; Ho, J.; Richardson, J.P.; Naglik, J.R. Some like it hot: Candida activation of inflammasomes. PLoS Pathog. 2020, 16, e1008975. [Google Scholar] [CrossRef]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef]
- Gomes, B.; Augusto, M.T.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol. Adv. 2018, 36, 415–429. [Google Scholar] [CrossRef]
- Struyfs, C.; Cammue, B.P.A.; Thevissen, K. Membrane-Interacting Antifungal Peptides. Front. Cell Dev. Biol. 2021, 9, 649875. [Google Scholar] [CrossRef]
- Donadu, M.G.; Peralta-Ruiz, Y.; Usai, D.; Maggio, F.; Molina-Hernandez, J.B.; Rizzo, D.; Bussu, F.; Rubino, S.; Zanetti, S.; Paparella, A. Colombian Essential Oil of Ruta graveolens against Nosocomial Antifungal Resistant Candida Strains. J. Fungi 2021, 7, 383. [Google Scholar] [CrossRef] [PubMed]
- Donadu, M.G.; Usai, D.; Marchetti, M.; Usai, M.; Mazzarello, V.; Molicotti, P.; Montesu, M.A.; Delogu, G.; Zanetti, S. Antifungal activity of oils macerates of North Sardinia plants against Candida species isolated from clinical patients with candidiasis. Nat. Prod. Res. 2020, 34, 3280–3284. [Google Scholar] [CrossRef] [PubMed]
- Bua, A.; Usai, D.; Donadu, M.G.; Delgado Ospina, J.; Paparella, A.; Chaves-Lopez, C.; Serio, A.; Rossi, C.; Zanetti, S.; Molicotti, P. Antimicrobial activity of Austroeupatorium inulaefolium (HBK) against intracellular and extracellular organisms. Nat. Prod. Res. 2018, 32, 2869–2871. [Google Scholar] [CrossRef] [PubMed]
- Dolan, K.; Montgomery, S.; Buchheit, B.; DiDone, L.; Wellingtone, M.; Krysan, D.J. Antifungal Activity of Tamoxifen: In Vitro and In Vivo Activities and Mechanistic Characterization. Antimicrob. Agents Chemother. 2009, 53, 3337–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koselny, K.; Green, J.; DiDone, L.; Halterman, J.P.; Fothergill, A.W.; Wiederhold, N.P.; Patterson, T.F.; Cushion, M.T.; Rappelye, C.; Wellington, M.; et al. The Celecoxib Derivative AR-12 Has Broad-Spectrum Antifungal Activity In Vitro and Improves the Activity of Fluconazole in a Murine Model of Cryptococcosis. Antimicrob. Agents Chemother. 2016, 60, 7115–7127. [Google Scholar] [CrossRef] [Green Version]
- Posch, W.; Wilflingseder, D.; Lass-Florl, C. Immunotherapy as an Antifungal Strategy in Immune Compromised Hosts. Curr. Clin. Microbiol. Rep. 2020, 7, 57–66. [Google Scholar] [CrossRef]
- Rachini, A.; Pietrella, D.; Lupo, P.; Torosantucci, A.; Chiani, P.; Bromuro, C.; Proietti, C.; Bistoni, F.; Cassone, A.; Vecchiarelli, A. An anti-beta-glucan monoclonal antibody inhibits growth and capsule formation of Cryptococcus neofonnans in vitro and exerts therapeutic, anticryptococcal activity in vivo. Infect. Immun. 2007, 75, 5085–5094. [Google Scholar] [CrossRef] [Green Version]
- Mambro, T.D.; Vanzolini, T.; Bruscolini, P.; Perez-Gaviro, S.; Marra, E.; Roscilli, G.; Bianchi, M.; Fraternale, A.; Schiavano, G.F.; Canonico, B.; et al. A new humanized antibody is effective against pathogenic fungi in vitro. Sci. Rep. 2021, 11, 19500. [Google Scholar] [CrossRef] [PubMed]
- Nosanchuk, J.D.; Dadachova, E. Radioimmunotherapy of fungal diseases: The therapeutic potential of cytocidal radiation delivered by antibody targeting fungal cell surface antigens. Front. Microbiol. 2012, 3, 283. [Google Scholar] [CrossRef] [Green Version]
- Heung, L.J.; Luberto, C.; Del Poeta, M. Role of sphingolipids in microbial pathogenesis. Infect. Immun. 2006, 74, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Levery, S.B.; Momany, M.; Lindsey, R.; Toledo, M.S.; Shayman, J.A.; Fuller, M.; Brooks, K.; Doong, R.L.; Straus, A.H.; Takahashi, H.K. Disruption of the glucosylceramide biosynthetic pathway in Aspergillus nidulans and Aspergillus fumigatus by inhibitors of UDP-Glc: Ceramide glucosyltransferase strongly affects spore germination, cell cycle, and hyphal growth (vol 525, pg 59, 2002). Febs Lett. 2002, 526, 151. [Google Scholar] [CrossRef] [Green Version]
- Rittershaus, P.C.; Kechichian, T.B.; Allegood, J.C.; Merrill, A.H., Jr.; Hennig, M.; Luberto, C.; Del Poeta, M. Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J. Clin. Investig. 2006, 116, 1651–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, S.M.; Thornton, R.A.; Frommer, B.R.; Curotto, J.E.; Rozdilsky, W.; Kurtz, M.B.; Giacobbe, R.A.; Bills, G.F.; Cabello, M.A.; Martín, I. The Discovery of Australifungin, a Novel Inhibitor of Sphinganine N-Acyltransferase from Sporormiella-Australis—Producing Organism, Fermentation, Isolation, and Biological-Activity. J. Antibiot. 1995, 48, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achenbach, H.; Mühlenfeld, A.; Fauth, U.; Zähner, H. The galbonolides. Novel, powerful antifungal macrolides from Streptomyces galbus ssp. eurythermus. Ann. N. Y. Acad. Sci. 1988, 544, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Mora, V.; Rellaa, A.; Farnouda, A.M.; Singha, A.; Munshia, M.; Bryana, A.; Naseema, S.; Konopkaa, J.B.; Ojimab, I.; Bullesbachc, E.; et al. Identification of a New Class of Antifungals Targeting the Synthesis of Fungal Sphingolipids. Mbio 2015, 6, e00647-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryštůfek, R.; Šácha, P.; Starková, J.; Brynda, J.; Hradilek, M.; Tloušt’ová, E.; Grzymska, J.; Rut, W.; Boucher, M.J.; Drąg, M.; et al. Re-emerging Aspartic Protease Targets: Examining Cryptococcus neoformans Major Aspartyl Peptidase 1 as a Target for Antifungal Drug Discovery. J. Med. Chem. 2021, 64, 6706–6719. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Clinically used antifungals grouped according to their mechanism of action with the most common agents in practice as examples.
Figure 1.
Clinically used antifungals grouped according to their mechanism of action with the most common agents in practice as examples.

Figure 2.
The chemical structures of amphotericin B (an example of a polyene); caspofungin (an example of an echinocandin); flucytosine; fluconazole (an example of an azole); and tavaborole.
Figure 2.
The chemical structures of amphotericin B (an example of a polyene); caspofungin (an example of an echinocandin); flucytosine; fluconazole (an example of an azole); and tavaborole.

Figure 3.
A summative figure showing the classification of new molecules as antifungals in clinical development.
Figure 3.
A summative figure showing the classification of new molecules as antifungals in clinical development.

Figure 4.
The chemical structures of (a) T-2307 and (b) pentamidine. The characteristic aryldiamidine moiety is highlighted in grey.
Figure 4.
The chemical structures of (a) T-2307 and (b) pentamidine. The characteristic aryldiamidine moiety is highlighted in grey.

Figure 5.
The chemical structures of (a) 1-[4-butylbenzyl]isoquinoline, (b) manogepix, and (c) fosmanogepix.
Figure 5.
The chemical structures of (a) 1-[4-butylbenzyl]isoquinoline, (b) manogepix, and (c) fosmanogepix.

Figure 6.
The chemical structures of (a) Nikkomycin Z and (b) Uridine Diphosphate (UDP)-N-acetyl glucosamine.
Figure 6.
The chemical structures of (a) Nikkomycin Z and (b) Uridine Diphosphate (UDP)-N-acetyl glucosamine.

Figure 7.
The chemical structures of olorofim.

Figure 8.
The chemical structures of (a) VL-2397 and (b) ferrichrome siderophore. To better visualize the structural resemblance, common fragments with ferrochrome siderophore are highlighted in grey.
Figure 8.
The chemical structures of (a) VL-2397 and (b) ferrichrome siderophore. To better visualize the structural resemblance, common fragments with ferrochrome siderophore are highlighted in grey.

Figure 9.
The chemical structure of Aureobasidin A.

Figure 10.
Chemical structures of (a) VT-1129, (b) VT-1161, and (c) VT-1598. Differences in chemical structures are highlighted in grey.
Figure 10.
Chemical structures of (a) VT-1129, (b) VT-1161, and (c) VT-1598. Differences in chemical structures are highlighted in grey.

Figure 11.
The chemical structures of (a) enfumafungin and (b) ibrexafungerp. The triterpenoid unit is shaded in grey.
Figure 11.
The chemical structures of (a) enfumafungin and (b) ibrexafungerp. The triterpenoid unit is shaded in grey.

Figure 12.
Hydrolysis reaction of the prodrug (a) isavuconazonium by mammalian plasma esterases to yield, (b) isavuconazole, and (c) by-product.
Figure 12.
Hydrolysis reaction of the prodrug (a) isavuconazonium by mammalian plasma esterases to yield, (b) isavuconazole, and (c) by-product.

Figure 13.
The chemical structures of (a) albaconazole and (b) iodiconazole.

Figure 14.
The chemical structures of (a) BSG005 and (b) nystatin. The structural modifications are shown in red.
Figure 14.
The chemical structures of (a) BSG005 and (b) nystatin. The structural modifications are shown in red.

Figure 15.
The chemical structures of (a) anidulafungin and (b) rezafungin. The chemical modification in the structure of rezafungin over anidulafungin is highlighted in grey.
Figure 15.
The chemical structures of (a) anidulafungin and (b) rezafungin. The chemical modification in the structure of rezafungin over anidulafungin is highlighted in grey.

Figure 16.
The chemical structure of the broad-spectrum chelator diethylenetriamine pentaacetate (DTPA).
Figure 16.
The chemical structure of the broad-spectrum chelator diethylenetriamine pentaacetate (DTPA).

Figure 17.
The chemical structures of (a) tamoxifen, (b) sertraline, and (c) AR-12.

Figure 18.
The chemical structures of (a) australifungin, (b) galbonolide A, (c) D-threo-PDMP, and (d) BHBM.
Figure 18.
The chemical structures of (a) australifungin, (b) galbonolide A, (c) D-threo-PDMP, and (d) BHBM.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The spectrum of activity, route of administration, production company, and current status of antifungals in clinical development. Clinical registration numbers were obtained from www.clinicaltrials.gov (accessed on 26 November 2021).
Table 1.
The spectrum of activity, route of administration, production company, and current status of antifungals in clinical development. Clinical registration numbers were obtained from www.clinicaltrials.gov (accessed on 26 November 2021).
| Agent | Spectrum of Activity | Route of Administration | Company/Sponsor | Current Status | Clinical Trial Registration Number |
|---|---|---|---|---|---|
| T-2307 | Broad spectrum | IV | Toyama Chemical Ltd. | Phase I completed (as stated in [57], but no actual data available in the literature) | Not available |
| Fosmanogepix | Candida spp. (except C. krusei) and Aspergillus spp. | PO/IV | Amplyx Pharmaceuticals | Phase II recruiting | NCT04240886 |
| Nikkomycin Z | Candida spp. and Aspergillus spp. | PO | University of Arizona | Phase I completed; however lack of funding and volunteers caused the termination of phase II studies | NCT00834184 |
| Olorofim | Aspergillus spp. and uncommon molds | PO/IV | F2G Ltd. | Phase II Phase III planned (not yet recruiting) | NCT03583164 NCT05101187 |
| VL-2397 | Aspergillus spp. and some Candida spp. | IV | Vical Biotechnology | No current development plans (phase II trial terminated early, because of a business decision) | NCT03327727 |
| Aureobasidin A | Board spectrum and proliferative tachyzoite form of toxoplasma (antiprotozoal) | PO/IV | Takara Bio Group | Preclinical | Not available |
| MGCD290 | Candida spp. and Aspergillus spp. | PO | MethylGene, Inc. | Further development suspended after phase II clinical trial | NCT01497223 |
| VT-1129 | Candida spp. and Cryptococus spp. | PO | Viamet Pharmaceuticals Inc. | Preclinical | Not available |
| VT-1161 | Candida spp., coccidioides spp., and Rhizopus spp. | PO | Mycovia Pharmaceuticals | Phase III completed | NCT03561701 |
| VT-1598 | Candida spp., Aspergillus spp., and Cryptococus spp. | PO | Mycovia Pharmaceuticals Clinical trial sponsored by National Institute of Allergy and Infectious Diseases (NIAID) | Phase I | NCT04208321 |
| Ibrexafungerp | Candida spp. and Aspergillus spp. | PO/IV | Scynexis, Inc. | Phase III | NCT03059992 |
| Isavuconazole | Broad spectrum | PO (Isavuconazonium sulfate PO/IV) | Basilea and Astellas Clinical trials sponsored by Memorial Sloan Kettering Cancer Center and M.D. Anderson Cancer Center, respectively. | Phase II trials completed FDA approved in 2015 for the treatment of aspergillosis and invasive mucormycosis | NCT03149055 NCT03019939 |
| Albaconazole | Broad spectrum | PO | Palau Pharma Clinical trial sponsored by GSK | Phase II completed | NCT00730405 |
| Iodiconazole | Broad spectrum | Topical | Second Military Medical University and Anhui Jiren Pharmaceutical | Phase III (as stated in [58], but no actual data available in the literature) | Not available |
| BSG005 | Broad spectrum | IV | Biosergen AS | Phase I | NCT04921254 |
| Rezafungin | Candida spp., Aspergillus spp., and Pneumocystis jirovecii | IV | Cidara Therapeutics, Inc. | Phase III | NCT03667690 NCT04368559 |
| SUBA-itraconazole | Broad spectrum | PO | Mayne Pharma Ltd. | Phase II FDA approved in 2018 for the treatment of aspergillosis, histoplasmosis, and blastomycosis in patients contraindicated to amphotericin B | NCT03572049 |
| Topical terbinafine solution | Onychomycosis | Topical 10% solution | Moberg Pharma | Phase III | NCT02859519 |
| Amphotericin B cochleate | Broad spectrum | PO | Matinas BioPharma | Phase II | NCT02629419 |
Table 2.
The general structures of some of the most recent (published in 2021), successful synthetic efforts from academia.
Table 2.
The general structures of some of the most recent (published in 2021), successful synthetic efforts from academia.
| Structural Type | General Structure * | Target | Ref. |
|---|---|---|---|
| Phloeodictine analogues |  | Fungal ergosterol biosynthesis | [59] |
| Biphenylethylaminoacetamides |  | Fungal cell wall integrity | [60] |
| Thiazoyl guanidine derivatives |  | Fungal ergosterol biosynthesis | [61] |
| Carboline HDAC inhibitors |  | Fungal histone deacetylase | [62] |
| Acyl hydrazides |  | Fungal fatty acid biosynthesis | [64] |
| Thiobenzoate scaffolds |  | Dual target: fungal acetohydroxyacid synthase and NLRP3 inflammasome | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bouz, G.; Doležal, M. Advances in Antifungal Drug Development: An Up-To-Date Mini Review. Pharmaceuticals 2021, 14, 1312. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121312
AMA Style
Bouz G, Doležal M. Advances in Antifungal Drug Development: An Up-To-Date Mini Review. Pharmaceuticals. 2021; 14(12):1312. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121312
Chicago/Turabian StyleBouz, Ghada, and Martin Doležal. 2021. "Advances in Antifungal Drug Development: An Up-To-Date Mini Review" Pharmaceuticals 14, no. 12: 1312. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121312
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.