Retinol-Containing Graft Copolymers for Delivery of Skin-Curing Agents

 , , and
, , and
Abstract
:1. Introduction
2. Experimental
2.1. Materials
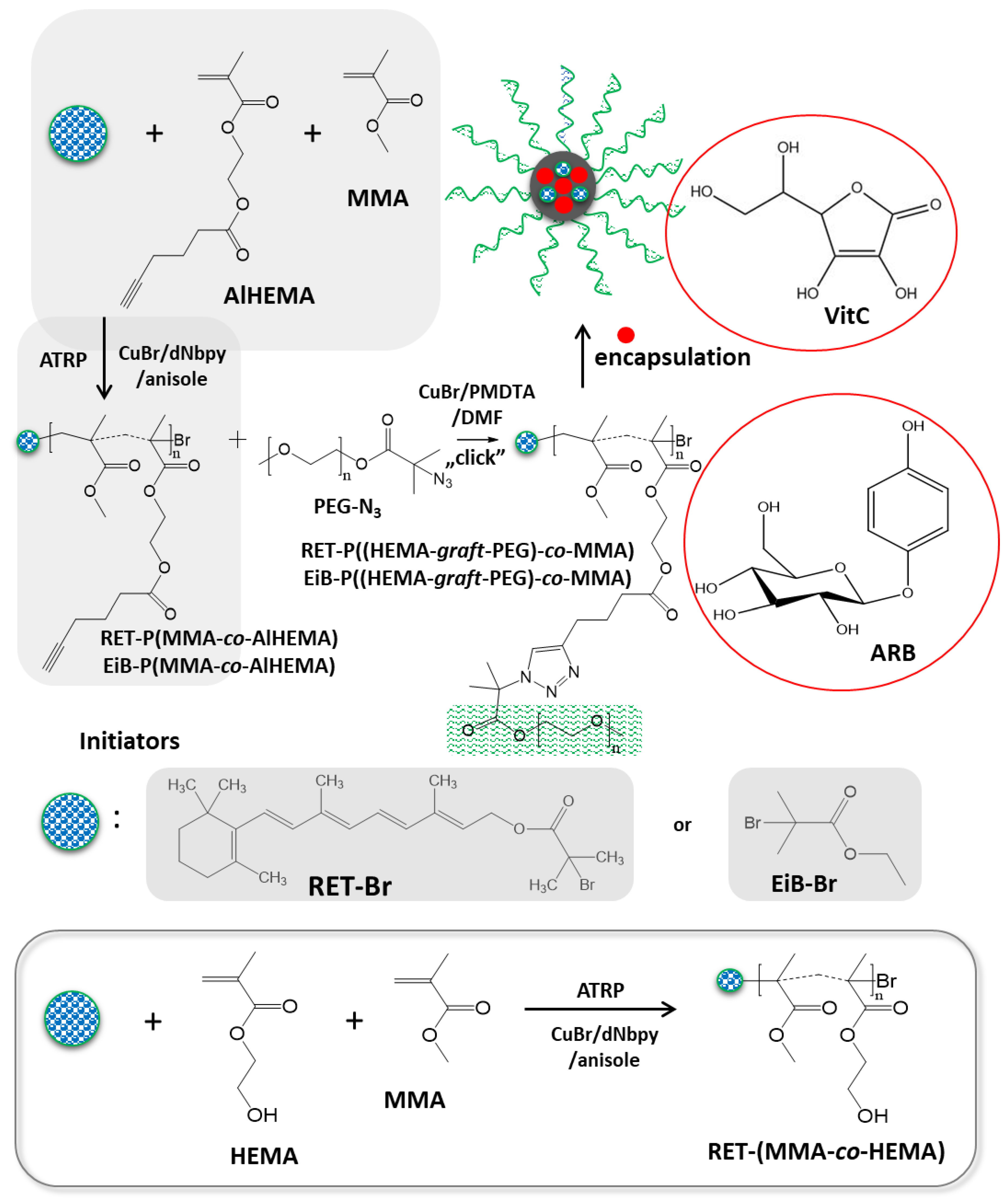
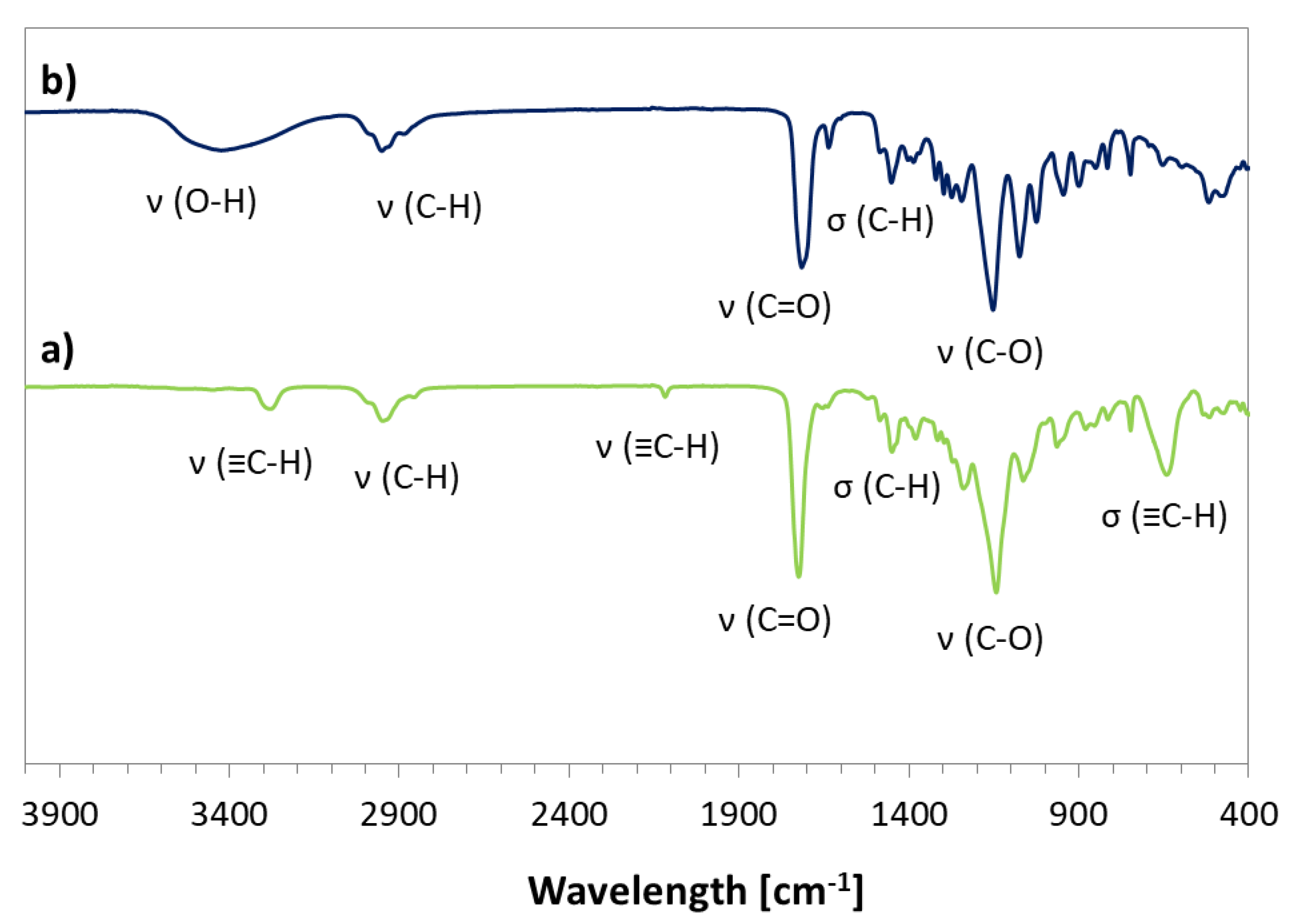
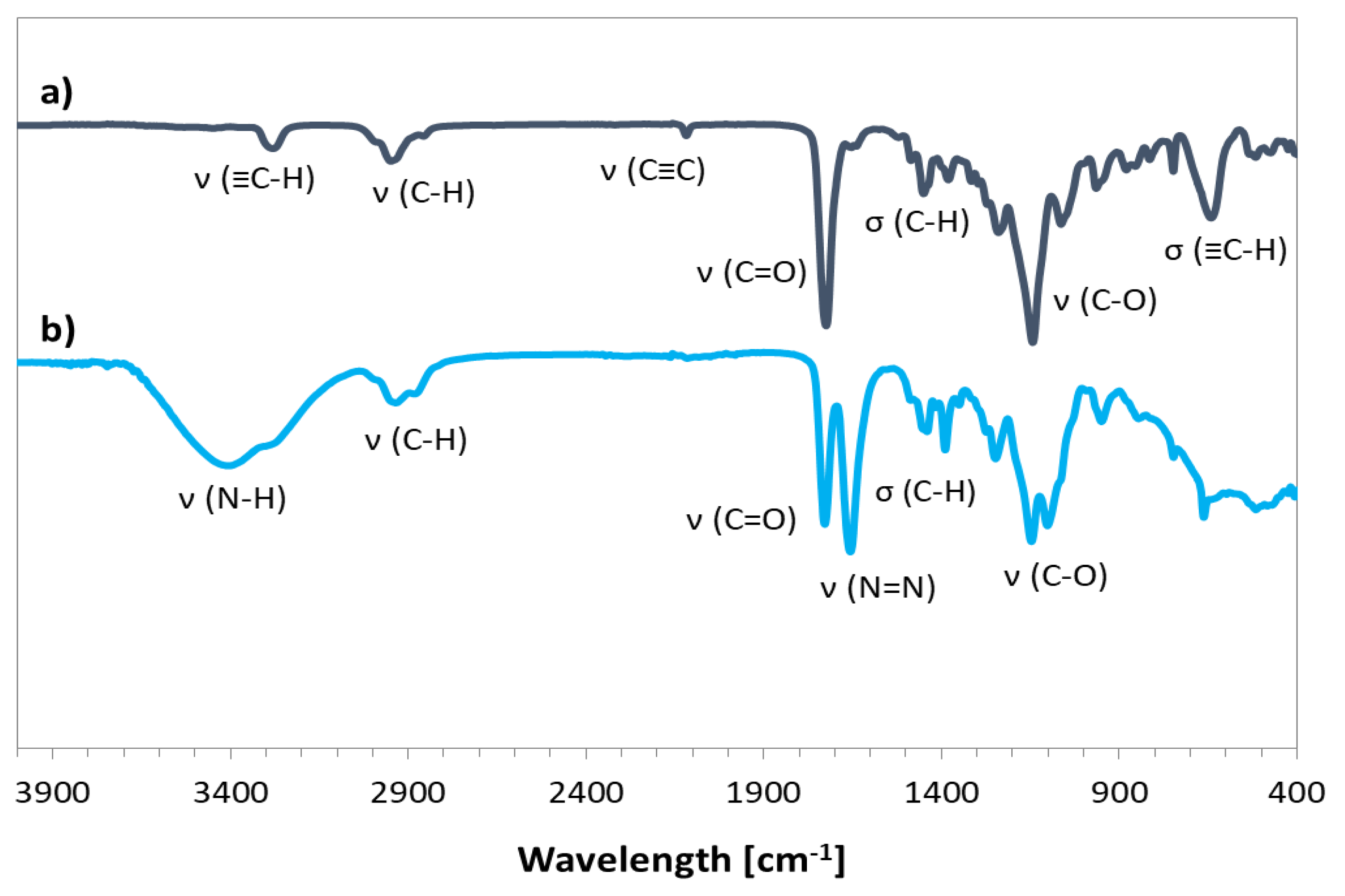
2.2. Synthesis of Alkyne-Functionalized HEMA (2-(Prop-1-En-2-Carbonyloxy)Ethyl Hex-5-Ynate, AlHEMA)
2.3. Synthesis of 2-Bromoisobutyrate Derivative of Retinol (3,7-Dimethyl-9-(2,6,6-Trimethylcyclohex-1-En-1-Yl)Nona-2,4,6,8-Tetraen-1-yl 2-Bromo-2-Methylpropanoate, RET-Br)
2.4. Synthesis of P(AlHEMA-co-MMA) with EiB-Br as Initiator (Example for III)
2.5. Synthesis of P(AlHEMA-co-MMA) with RET-Br as Initiator (Example for VI)
2.6. Synthesis of P(HEMA-co-MMA) (VII–IX)
2.7. Synthesis of Poly(Ethylene Glycol)Methyl Ether 2-Azidoisobutyrate (PEG-N3)
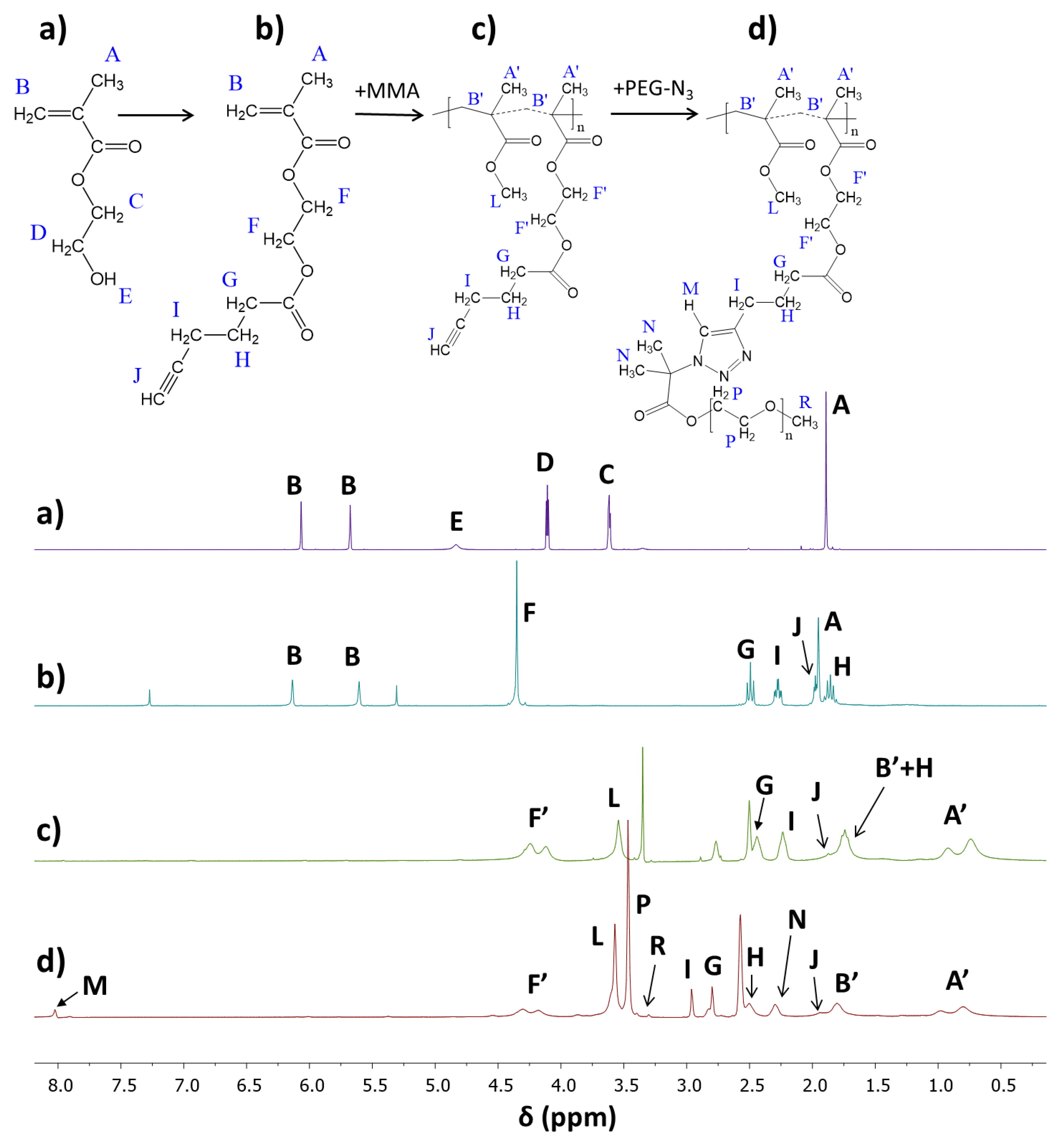
2.8. “Click” Chemistry Azide–Alkyne Reactions (Example for IVc)
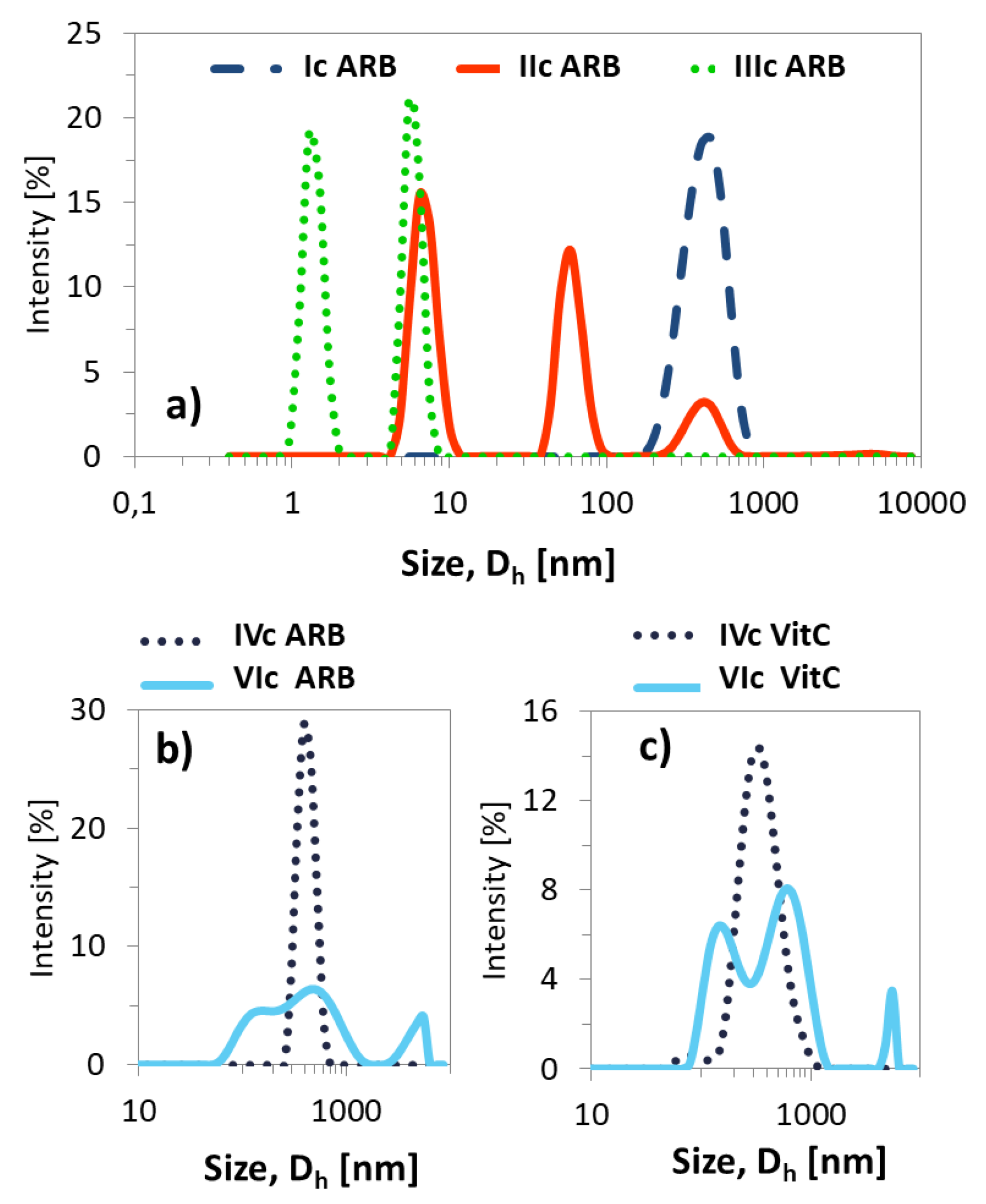
2.9. Incorporation of Active Substance into Polymeric Micelles
2.10. Active Substance Release Studies
2.11. Characterization
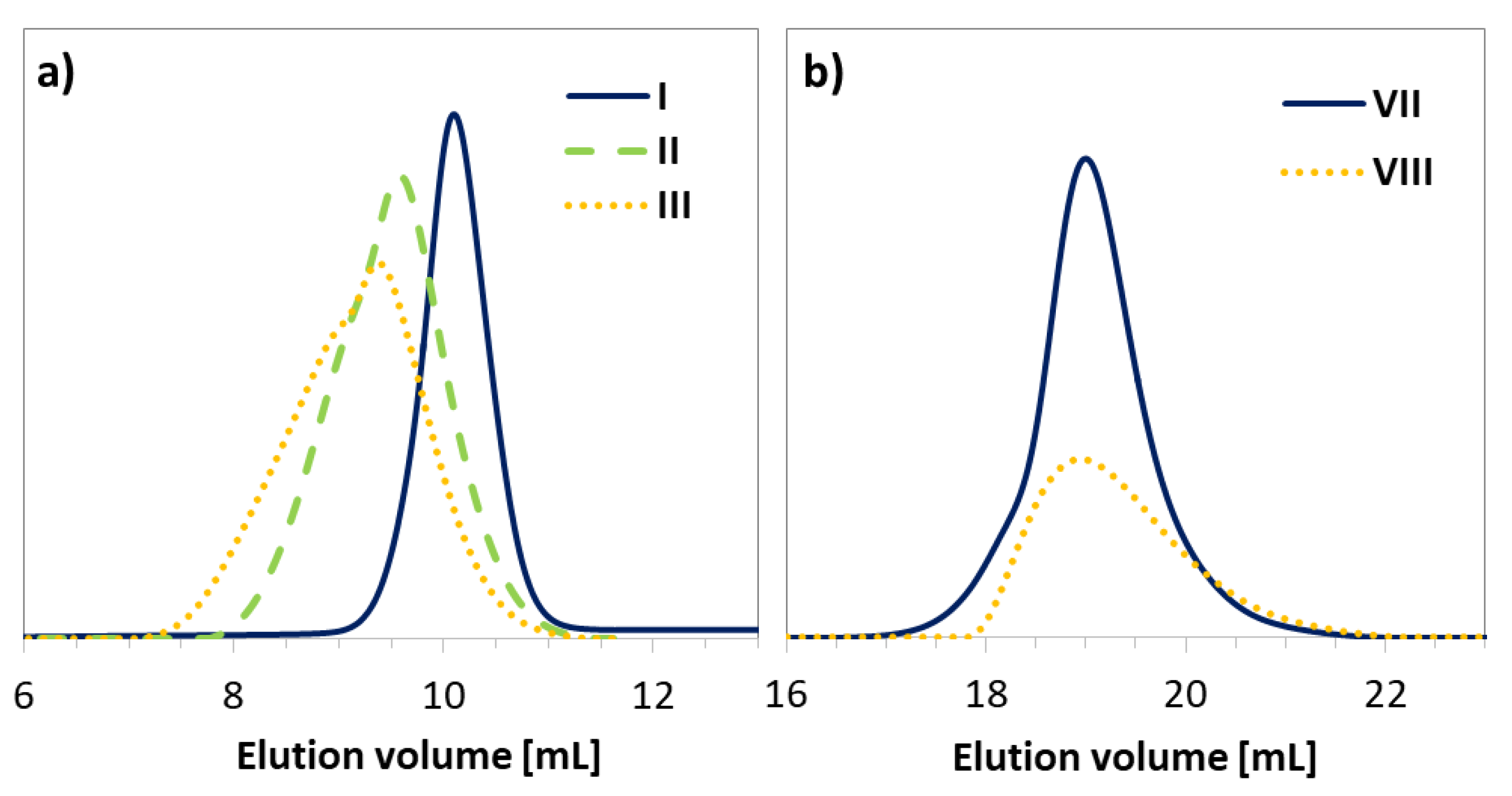
3. Results and Discussion
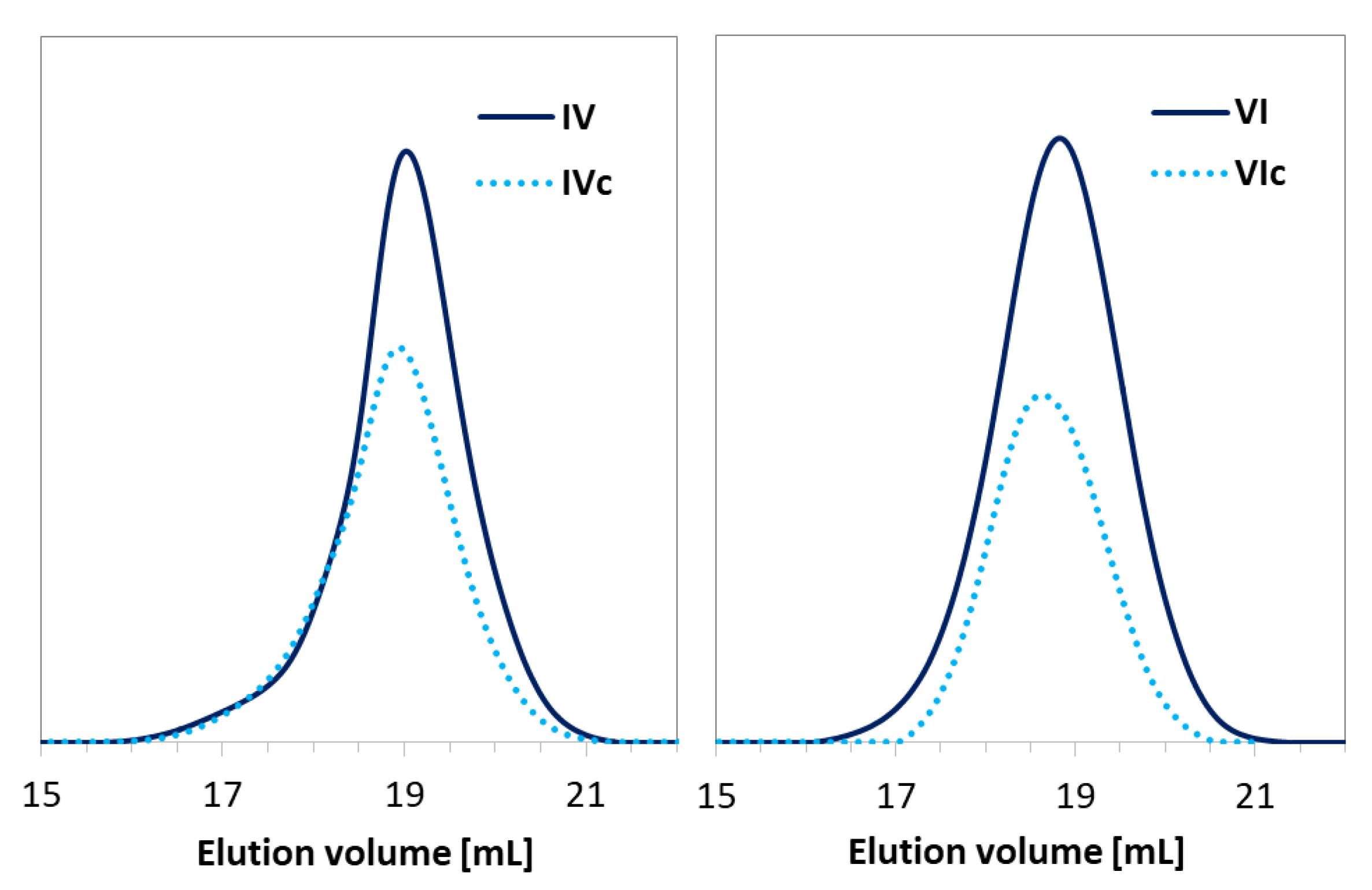
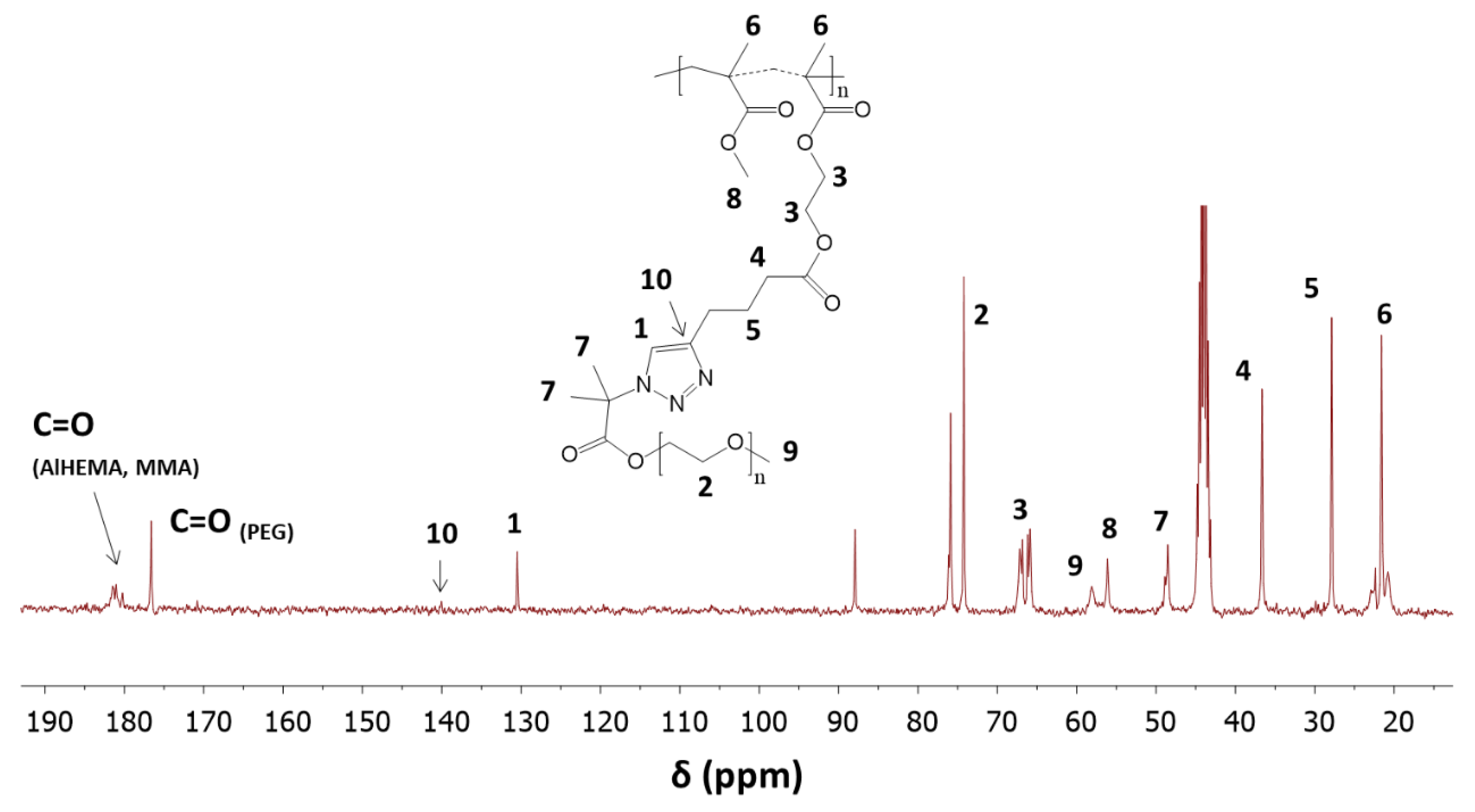
Click Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xu, X.; Ho, W.; Zhang, X.; Bertrand, N.; Farokhzad, O. Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 2015, 21, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Trase, I.; Ren, M.; Duval, K.; Guo, X.; Chen, Z. Design of Nanoparticle-Based Carriers for Targeted Drug Delivery. J. Nanomater. 2016, 2016, 1087250. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Kamaly, N.; Memic, A.; Shafiee, H. Self-assembled peptide-based nanostructures: Smart nanomaterials toward targeted drug delivery. Nano Today 2016, 11, 41–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodzinski, A.; Guduru, R.; Liang, P.; Hadjikhani, A.; Stewart, T.; Stimphil, E.; Runowicz, C.; Cote, R.; Altman, N.; Datar, R.; et al. Targeted and controlled anticancer drug delivery and release with magnetoelectric nanoparticles. Sci. Rep. 2016, 6, 20867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Yadav, T.; Sharma, S.; Nayak, A.; Kumari, A.; Mishra, N. Polymers in Drug Delivery. J. Biosci. Med. 2016, 4, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Mady, M.M.; Darwish, M.M.; Khalil, S.; Khalil, W.M. Biophysical studies on chitosan-coated liposomes. Eur. Biophys. J. 2009, 38, 1127–1133. [Google Scholar] [CrossRef]
- Prado, A.G.S.; Santos, A.L.F.; Pedroso, C.P.; Carvalho, T.O.; Braga, L.R.; Evangelista, S.M. Vitamin A and vitamin E interaction behavior on chitozan microspheres. J. Therm. Anal. Calorim. 2011, 16, 415–420. [Google Scholar] [CrossRef]
- Creuzet, C.; Kadi, S.; Rinaudo, M.; Auze´ly-Velty, R. New associative systems based on alkylated hyaluronic acid. Synthesis and aqueous solution properties. Polymer 2006, 47, 2706–2713. [Google Scholar] [CrossRef]
- Olejnik, A.; Goscianska, J.; Zielinska, A.; Nowak, I. Stability determination of the formulations containing hyaluronic acid. Int. J. Cosmet. Sci. 2015, 37, 401–407. [Google Scholar] [CrossRef]
- Avila Rodriguez, M.I.; Rodriguez Barroso, L.G.; Sanchez, M.L. Collagen: A review on its sources and potential cosmetic applications. J. Cosmet. Dermatol. 2018, 17, 20–26. [Google Scholar] [CrossRef]
- Parisi, O.I.; Malivindi, R.; Amone, F.; Ruffo, M.; Malanchin, R.; Carlomagno, F.; Piangiolino, C.; Nobile, V.; Pezzi, V.; Scrivano, L.; et al. Safety and efficacy of dextran-rosmarinic acid conjugates as innovative polymeric antioxidants in skin whitening: What Is the Evidence? Cosmetics 2017, 4, 28. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Zhang, X.-Z.; Cheng, H.; Zhu, J.-J.; Cheng, S.-X.; Zhuo, R.-X. Self-Assembled, Thermosensitive PCL-g-P(NIPAAm-co-HEMA) Micelles for Drug Delivery. Macromol. Rapid Commun. 2006, 27, 1913–1919. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.D.; Wu, D.Q.; Zhang, X.Z.; Zhuo, R.X. Synthesis of thermosensitive P(NIPAAm-co-HEMA)/cellulose hydrogels via. ‘‘click” chemistry. Carbohydr. Polym. 2009, 77, 583–589. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Zhao, B.; Li, Z.D.; Lin, W.J.; Zhang, C.Y.; Guo, X.D.; Wang, J.F.; Zhang, L.J. PH-sensitive micelles self-assembled from multi-arm star triblock-co-polymers poly(e-caprolactone)-b-poly(2-(diethylamino)ethylmethacrylate)-b-poly(poly(ethylene glycol)methyl ether methacrylate) for controlled anticancer drug delivery. Acta Biomater. 2013, 9, 7679–7690. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.L.; Boker, A.; Zhang, M.F.; Krausch, G.; Muller, A.H.E. Amphiphilic cylindrical core-shell brushes via. a "grafting from" process using ATRP. Macromolecules 2001, 34, 6883–6888. [Google Scholar] [CrossRef]
- Neugebauer, D.; Odrobińska, J.; Bielas, R.; Mielańczyk, A. Design of systems based on 4-armed star-shaped polyacids for indomethacin delivery. New J. Chem. 2016, 40, 10002–10011. [Google Scholar] [CrossRef]
- Chang, L.L.; Liu, J.J.; Zhang, J.H.; Deng, L.D.; Dong, A.J. PH-sensitive nanoparticles prepared from amphiphilic and biodegradable methoxy poly(ethylene glycol)-block-(polycaprolactone-graft-poly(methacrylic acid)) for oral drug delivery. Polym. Chem. 2013, 4, 1430–1438. [Google Scholar] [CrossRef]
- Ruan, G.; Feng, S.S. Preparation and characterization of poly(lactic acid)–poly(ethylene glycol)–poly(lactic acid) (PLA–PEG–PLA) microspheres for controlled release of paclitaxel. Biomaterials 2003, 24, 5037–5044. [Google Scholar] [CrossRef]
- Maksym-Bebenek, P.; Neugebauer, D. Study on Self-Assembled Well-Defined PEG Graft Copolymers as Efficient Drug-Loaded Nanoparticles for Anti-Inflammatory Therapy. Macromol. Biosci. 2015, 15, 1616–1624. [Google Scholar] [CrossRef]
- Ishizu, K.; Satoh, J.; Sogabe, A. Architecture and solution properties of AB-type brush-block-brush amphiphilic copolymers via ATRP techniques. J. Colloid Interface Sci. 2004, 274, 472–479. [Google Scholar] [CrossRef]
- Wolf, F.; Friedemann, N.; Frey, H. Poly(lactide)-block-Poly(HEMA) Block Copolymers: An Orthogonal One-Pot Combination of ROP and ATRP, Using a Bifunctional Initiator. Macromolecules 2009, 42, 5622–5628. [Google Scholar] [CrossRef]
- Huang, L.-M.; Li, L.-D.; Shang, L.; Zhou, Q.-H.; Lin, J. Preparation of pH-sensitive micelles from miktoarm star block copolymers by ATRP and their application as drug nanocarriers. React. Funct. Polym. 2016, 107, 28–34. [Google Scholar] [CrossRef]
- Cheng, R.; Wang, X.; Chen, W.; Meng, F.; Deng, C.; Liu, H.; Zhong, Z. Biodegradable poly(3-caprolactone)-g-poly(2-hydroxyethyl methacrylate) graft copolymer micelles as superior nano-carriers for ‘‘smart’’ doxorubicin release. J. Mater. Chem. 2012, 22, 11730–11738. [Google Scholar] [CrossRef]
- Gromadzki, D.; Stepanek, P.; Makuska, R. Synthesis of densely grafted copolymers with tert-butyl methacrylate/2-(dimethylamino ethyl) methacrylate side chains as precursors for brush polyelectrolytes and polyampholytes. Mater. Chem. Phys. 2013, 137, 709–715. [Google Scholar] [CrossRef]
- Bury, K.; Du Prez, F.; Neugebauer, D. Self-assembling linear and star shaped poly(ε-caprolactone)/poly[(meth)acrylic acid] block copolymers as carriers of indomethacin and quercetin. Macromol. Biosci. 2013, 13, 1520–1530. [Google Scholar] [CrossRef]
- Neugebauer, D. Two decades of molecular brushes by ATRP. Polymer 2015, 72, 413–421. [Google Scholar] [CrossRef]
- Neugebauer, D.; Bury, K.; Biela, T. Novel Hydroxyl-Functionalized Caprolactone Poly(meth)acrylates Decorated with tert-Butyl Groups. Macromolecules 2012, 45, 4989–4996. [Google Scholar] [CrossRef]
- Maksym-Bębenek, P.; Biela, T.; Neugebauer, D. Water soluble well-defined acidic graft copolymers based on a poly(propylene glycol) macromonomer. RSC Adv. 2015, 5, 3627–3635. [Google Scholar] [CrossRef]
- Cai, Y.L.; Hartenstein, M.; Muller, A.H.E. Synthesis of amphiphilic graft copolymers of n-butyl acrylate and acrylic acid by atom transfer radical copolymerization of macromonomers. Macromolecules 2004, 37, 7484–7490. [Google Scholar] [CrossRef]
- Neugebauer, D.; Zhang, Y.; Pakula, T.; Sheiko, S.S.; Matyjaszewski, K. Densely-Grafted and Double-Grafted PEO Brushes via ATRP. A Route to Soft Elastomers. Macromolecules 2003, 36, 6746–6755. [Google Scholar] [CrossRef]
- Neugebauer, D.; Zhang, Y.; Pakula, T.; Matyjaszewski, K. Heterografted PEO-PnBA brush copolymers. Polymer 2003, 44, 6863–6871. [Google Scholar] [CrossRef]
- Neugebauer, D. Modifications of Hydroxyl-Functionalized HEA/HEMA and Their Polymers in the Synthesis of Functional and Graft Copolymers. Curr. Org. Synth. 2017, 14, 1–12. [Google Scholar] [CrossRef]
- Montheard, J.P.; Chatzopoulos, M.; Chappard, D. 2-Hydroxyethyl Methacrylate (HEMA): Chemical Properties and Applications in Biomedical Fields. J. Macromol. Sci. C 1992, 32, 1–34. [Google Scholar] [CrossRef]
- Passos, M.F.; Dias, D.R.C.; Bastos, G.N.T.; Jardini, A.L.; Benatti, A.C.B.; Dias, C.G.B.T.; Filho, R.M. PHEMA hydrogels. Synthesis, kinetics and in vitro tests. J. Therm. Anal. Calorim. 2016, 125, 361–368. [Google Scholar] [CrossRef]
- Li, C.C.; Chauhan, A. Ocular transport model for ophthalmic delivery of timolol through p-HEMA contact lenses. J. Drug Deliv. Sci. Technol. 2007, 17, 69–79. [Google Scholar] [CrossRef]
- Chirila, T.V.; Constable, I.J.; Crawford, G.J.; Vijayasekaran, S.; Thompson, D.E.; Chen, Y.-C.; Fletcher, W.A.; Griffin, B.J. Poly(2-hydroxyethyl methacrylate) sponges as implant materials: In Vivo and In Vitro evaluation of cellular invasion. Biomaterials 1993, 14, 26–38. [Google Scholar] [CrossRef]
- Ling, J.; Zheng, Z.; Köhler, A.; Müller, A.H.E. Rod-Like Nano-Light Harvester. Macromol. Rapid Commun. 2014, 35, 52–55. [Google Scholar] [CrossRef]
- Beers, K.L.; Gaynor, S.G.; Matyjaszewski, K.; Sheiko, S.S.; Moller, M. The Synthesis of Densely Grafted Copolymers by Atom Transfer Radical Polymerization. Macromolecules 1998, 31, 9413–9415. [Google Scholar] [CrossRef]
- Sumerlin, B.S.; Tsarevsky, N.V.; Louche, G.; Lee, R.Y.; Matyjaszewski, K. Highly efficient “click” functionalization of poly(3-azidopropyl methacrylate) prepared by ATRP. Macromolecules 2005, 38, 7540–7545. [Google Scholar] [CrossRef]
- Mishra, V.; Jung, S.-H.; Park, J.M.; Jeong, H.M.; Lee, H.-I. Triazole-Containing Hydrogels for Time-Dependent Sustained Drug Release. Macromol. Rapid Commun. 2014, 35, 442–446. [Google Scholar] [CrossRef]
- Erol, F.E.; Sinirlioglu, D.; Cosgun, S.; Muftuoglu, A.E. Synthesis of Fluorinated Amphiphilic Block Copolymers Based on PEGMA, HEMA, and MMA via ATRP and CuAAC Click Chemistry. Int. J. Polym. Sci. 2014, 2014, 464806. [Google Scholar] [CrossRef]
- Bach, L.G.; Islam, R.; Nga, T.T.; Binh, M.T.; Hong, S.S.; Gal, Y.S.; Taek, L.K. Chemical Modification of Polyhedral Oligomeric Silsesquioxanes by Functional Polymer via. Azide-Alkyne Click Reaction. J. Nanosci. Nanotechnol. 2013, 13, 1970–1973. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Matyjaszewski, K. Synthesis of Molecular Brushes by “Grafting onto” Method: Combination of ATRP and Click Reactions. J. Am. Chem. Soc. 2007, 129, 6633–6639. [Google Scholar] [CrossRef] [PubMed]
- Odrobińska, J.; Neugebauer, D. Retinol derivative as bioinitiator in the synthesis of hydroxyl-functionalized polymethacrylates for micellar delivery systems. Express Polym. Lett. 2019, 13, 806–817. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M1/M2 | Time (h) | Conversion (%) | DPn,GC | Mn,GC (g/mol) | Mn a (g/mol) | a | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| NMR | GC | |||||||||
| M1 | M2 | M1 | M2 | |||||||
| I | 25/75 | 4.0 | 16 | 28 | 34 | 27 | 115 | 15,900 | 21,400 | 1.36 |
| II | 50/50 | 4.5 | 48 | 50 | 47 | 52 | 198 | 31,600 | 33,600 | 2.06 |
| III | 75/25 | 4.5 | 46 | 45 | 42 | 47 | 173 | 33,100 | 35,500 | 1.83 |
| IV | 25/75 | 24 | 33 | 29 | 23 | 22 | 89 | 12,200 | 25,400 | 1.68 |
| V | 50/50 | 24 | 27 | 15 | 18 | 17 | 71 | 12,000 | 17,800 | 1.72 |
| VI | 75/25 | 24 | 32 | 24 | 26 | 30 | 109 | 21,000 | 30,400 | 1.65 |
| DPAlHEMA | FAlHEMA (%) | Eclick (%) | ntriazole | DG (%) | Fhydrophilic (wt%) | Mn,NMR (g/mol) | Mn,GPC a (g/mol) | a | CMC (mg/mL) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Ic | 34 | 30 | 33 | 11 | 10 | 47 | 28,900 | 58,600 | 3.40 | 0.0159 |
| IIc | 94 | 47 | 31 | 29 | 15 | 54 | 69,300 | 24,100 | 1.34 | 0.0169 |
| IIIc | 126 | 73 | 27 | 34 | 20 | 57 | 77,300 | 36,500 | 1.50 | 0.0229 |
| IVc | 23 | 26 | 21 | 5 | 6 | 35 | 17,200 | 29,000 | 1.62 | 0.0836 |
| VIc | 79 | 72 | 64 | 51 | 47 | 76 | 75,000 | 35,900 | 1.42 | 0.4283 |
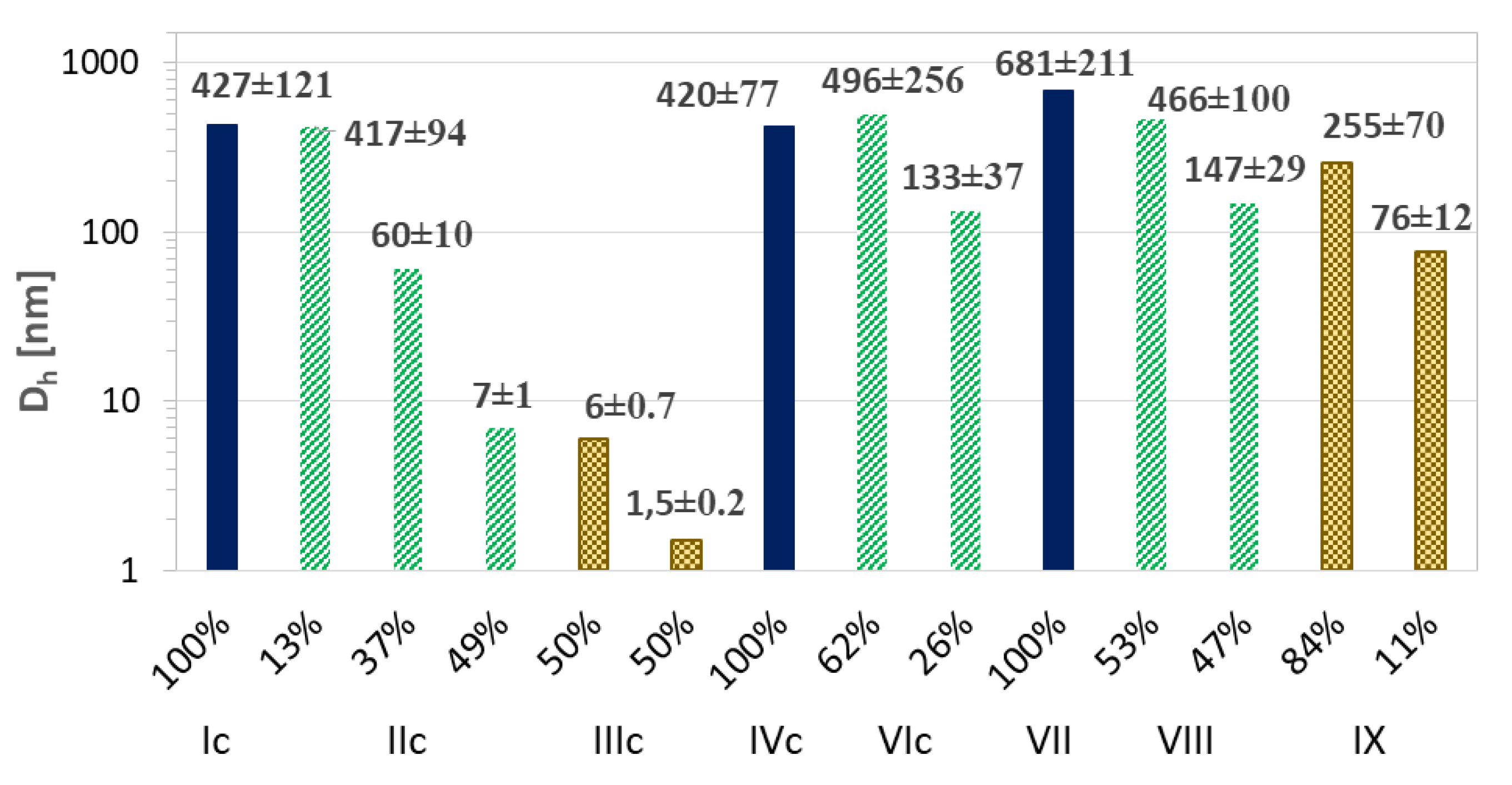
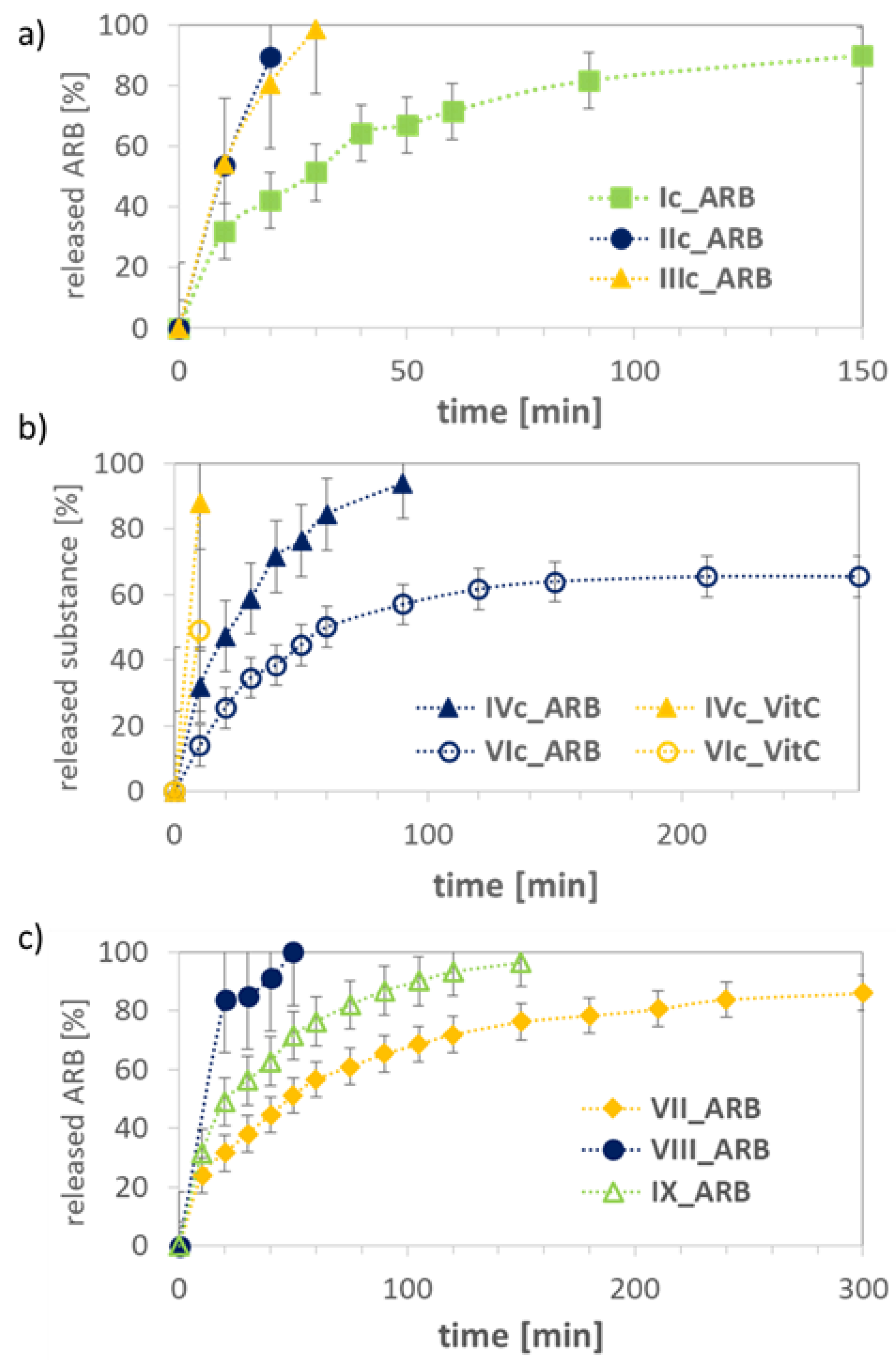
| Dh a (nm) | PDI | DLC (%) | Maximum Amount of Released Drug (%)/Time (h) | |||||
|---|---|---|---|---|---|---|---|---|
| ARB | VitC | ARB | VitC | ARB | VitC | ARB | VitC | |
| Ic | 427 | - | 0.508 ± 0.028 | - | 64 | - | 90/2.5 | - |
| IIc | 80 b | - | 0.717 ± 0.018 | - | 49 | - | 90/0.3 | - |
| IIIc | 7 b | - | 0.983 ± 0.016 | - | 56 | - | 99/0.5 | - |
| IVc | 420 | 369 | 0.241 ± 0.046 | 0.200 ± 0.063 | 99 | 16 | 94/1.5 | 88/0.17 |
| VIc | 310 b | 377 b | 0.634 ± 0.007 | 0.621 ± 0.085 | 87 | 13 | 65/4.5 | 49/0.17 |
| VII | 681 | 834 c | 0.093 ± 0.053 | 0.261 ± 0.081 | 55 | 87 c | 86/5.0 | 62/1.0 c |
| VIII | 316 b | 250 c | 0.904 ± 0.002 | 0.281 ± 0.060 | 48 | 78 c | 100/0.8 | 48/1.0 c |
| IX | 222 b | - | 0.691 ± 0.086 | - | 75 | - | 96/2.5 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odrobińska, J.; Niesyto, K.; Erfurt, K.; Siewniak, A.; Mielańczyk, A.; Neugebauer, D. Retinol-Containing Graft Copolymers for Delivery of Skin-Curing Agents. Pharmaceutics 2019, 11, 378. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11080378
Odrobińska J, Niesyto K, Erfurt K, Siewniak A, Mielańczyk A, Neugebauer D. Retinol-Containing Graft Copolymers for Delivery of Skin-Curing Agents. Pharmaceutics. 2019; 11(8):378. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11080378
Chicago/Turabian StyleOdrobińska, Justyna, Katarzyna Niesyto, Karol Erfurt, Agnieszka Siewniak, Anna Mielańczyk, and Dorota Neugebauer. 2019. "Retinol-Containing Graft Copolymers for Delivery of Skin-Curing Agents" Pharmaceutics 11, no. 8: 378. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics11080378