Protective Efficacy of Inhaled BCG Vaccination Against Ultra-Low Dose Aerosol M. tuberculosis Challenge in Rhesus Macaques

 , , , ,
, , , ,
Abstract
:1. Introduction
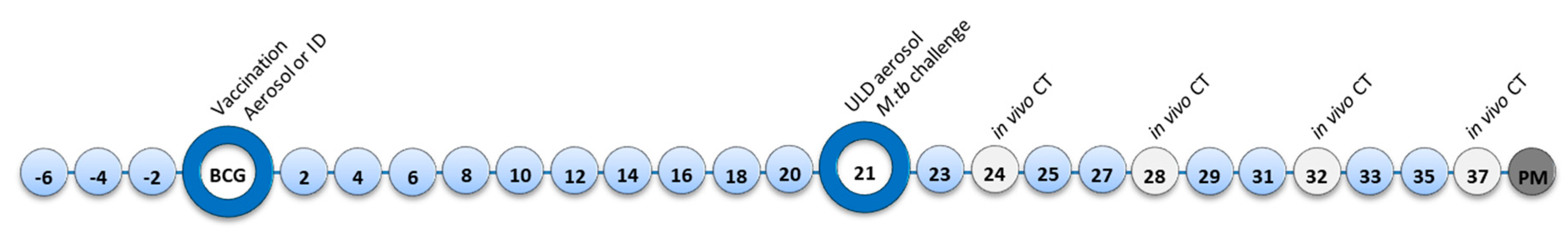
2. Materials and Methods
2.1. Experimental Animals
2.2. Vaccination
2.3. M. Tuberculosis Challenge Strain
2.4. Aerosol Exposure
2.4.1. Apparatus and Procedure
2.4.2. Quantification of Ultra-Low Aerosol Dose
2.5. Computed Tomography (CT) Imaging
2.6. Clinical Assessment
2.7. Immune Response Analysis
2.7.1. Interferon-Gamma (IFN-γ) ELISpot
2.7.2. Quantification of Secreted Biomarkers in Mycobacterial Antigen Stimulated Blood Cultures
2.8. Intracellular Cytokine Staining and Memory T-cell Analysis
2.8.1. Polyfunctional Intracellular Cytokine Staining and Antigen-Specific Memory T-cell Assay
2.8.2. Flow Cytometric Acquisition and Analysis
2.9. Necropsy
2.10. Pathology Studies
2.11. Histological Examination
2.12. Bacteriology
2.13. Statistical Analyses
3. Results
3.1. Safety of ID and Aerosol Delivered BCG
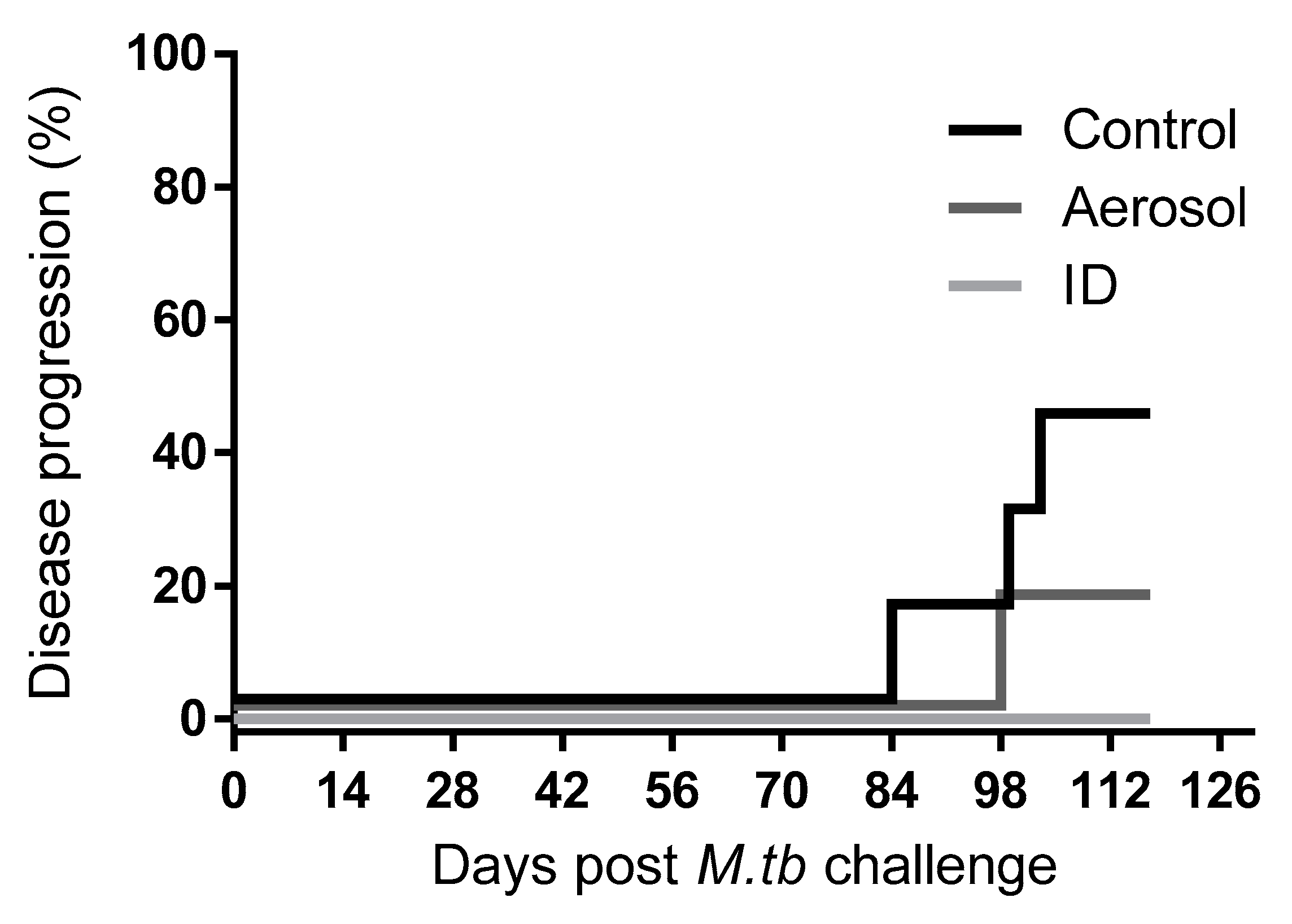
3.2. M. Tuberculosis Challenge Dose and Disease Progression
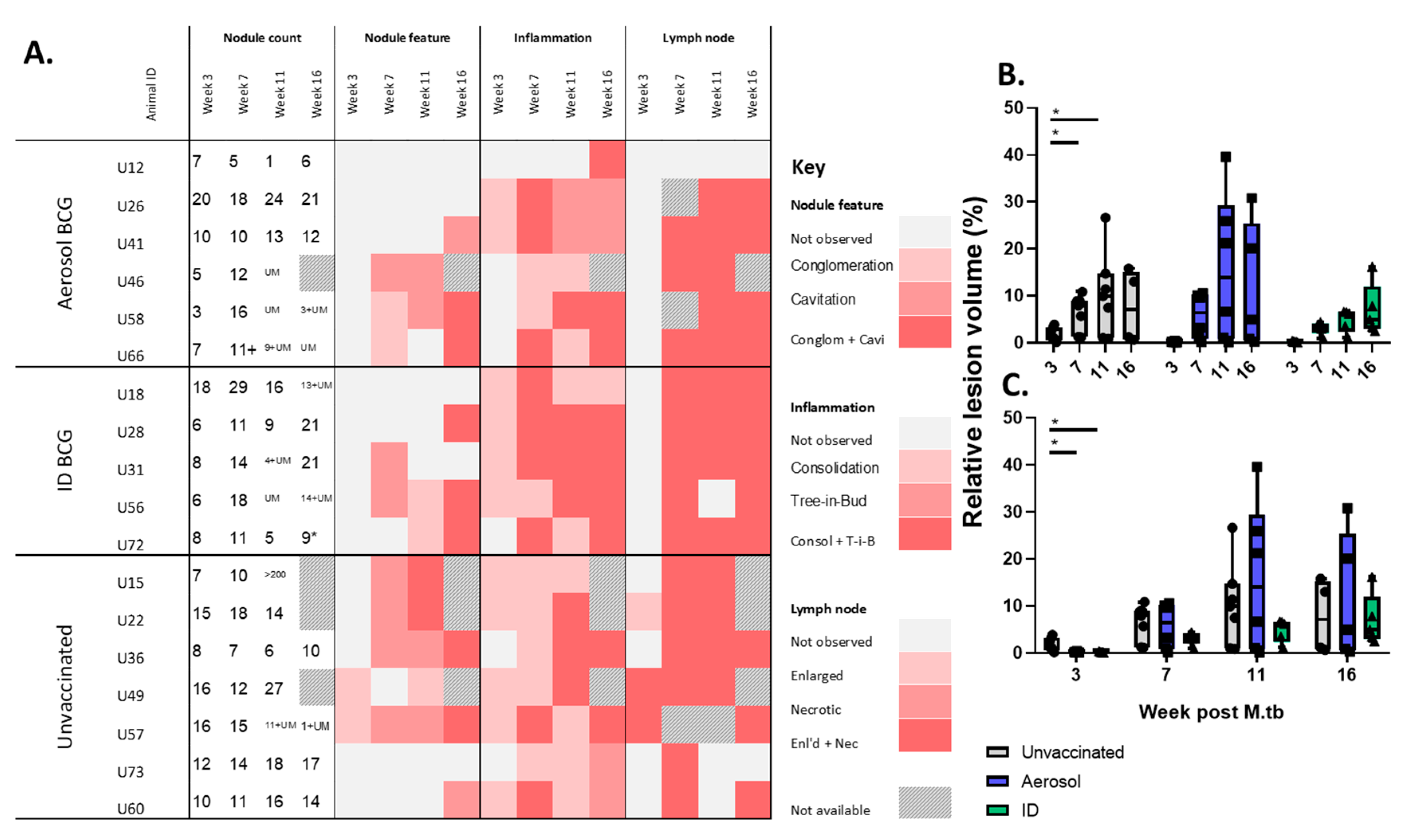
3.3. In Life CT Evaluation of Disease Pathology
3.4. Disease Pathology Recorded in Pulmonary and Extra-Pulmonary Tissues
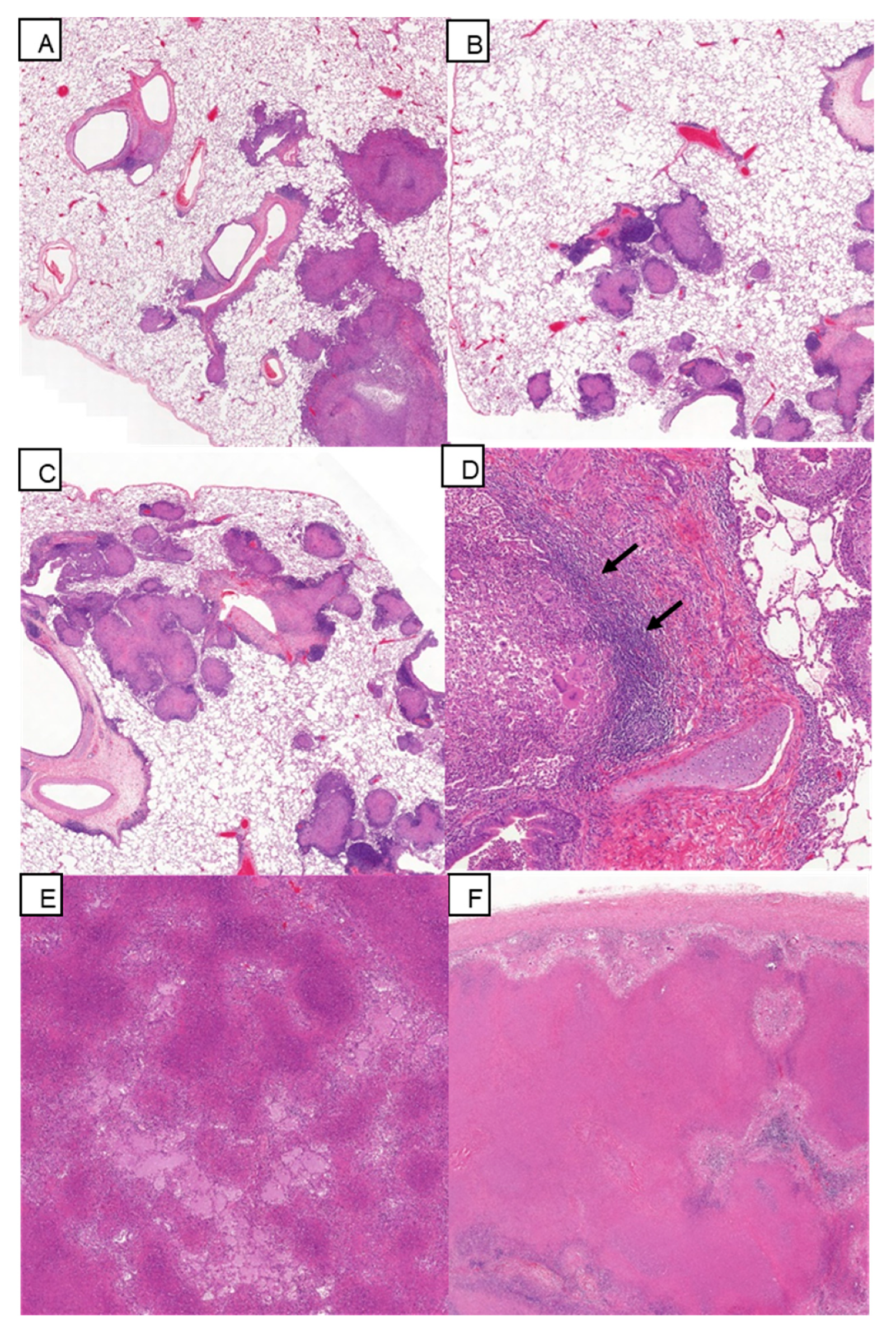
3.5. Histopathology
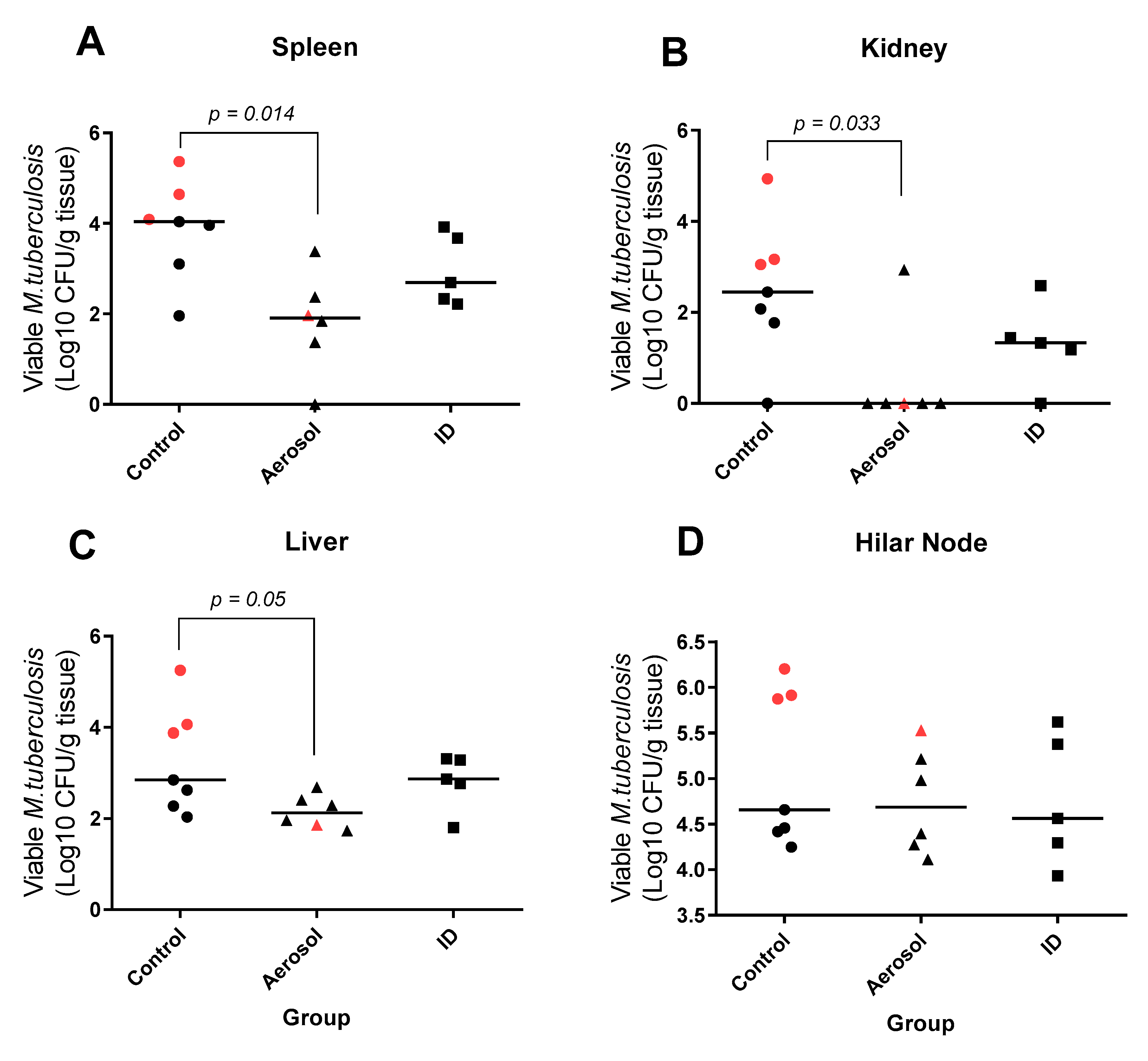
3.6. Viable M. Tuberculosis Recovered from Extra-Pulmonary Tissues
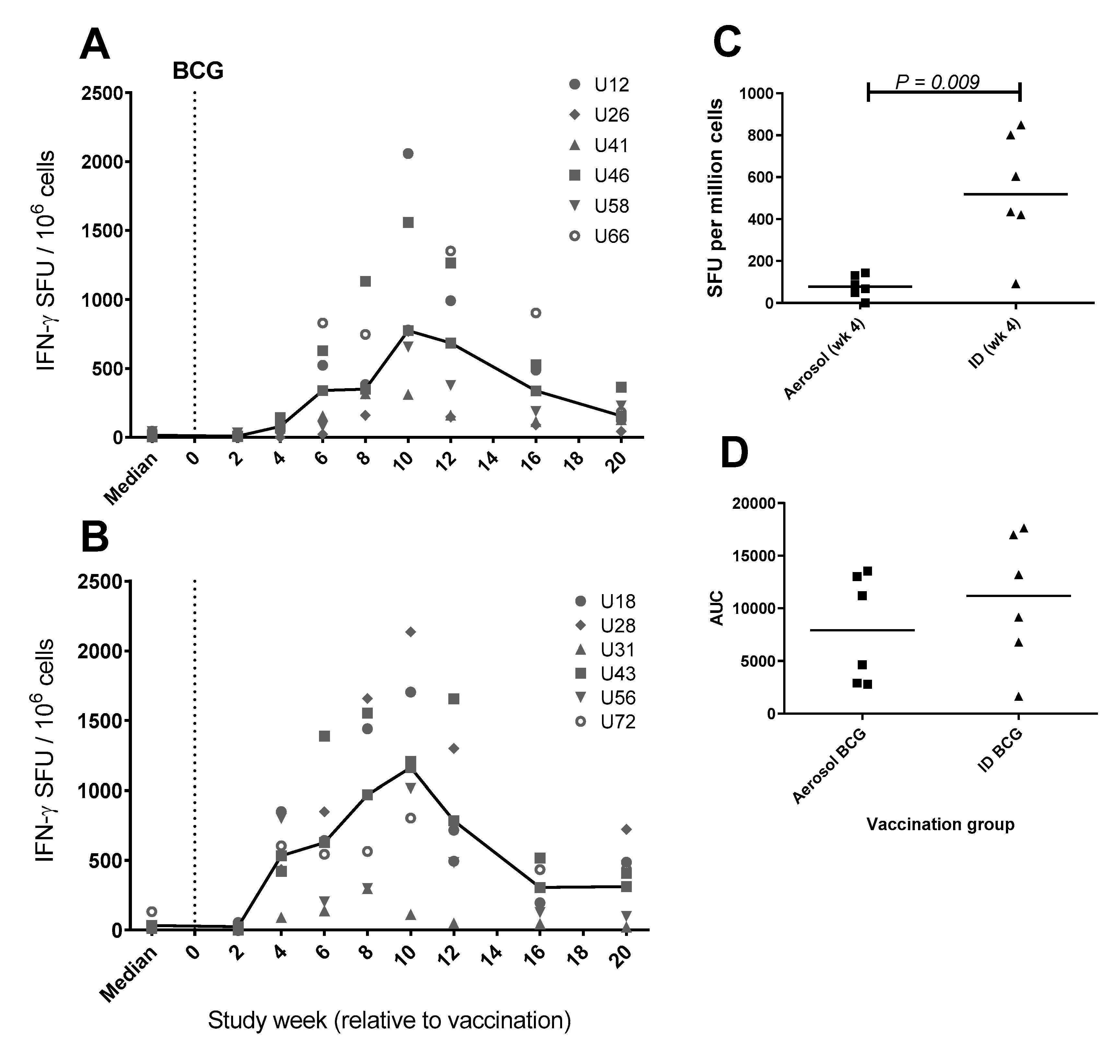
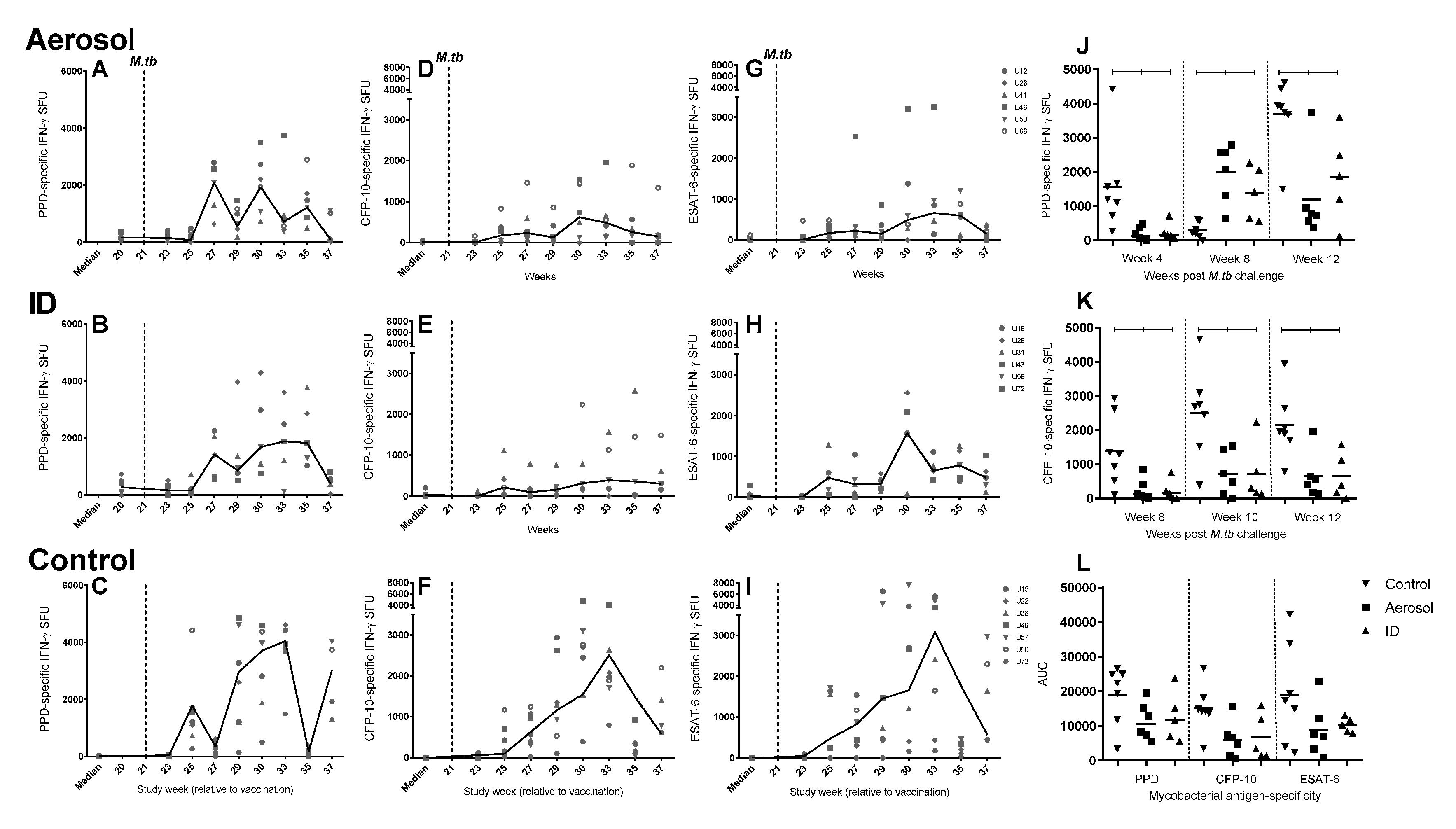
3.7. Frequency of Antigen-Specific IFN-γ Secreting Cells Measured by ELISpot
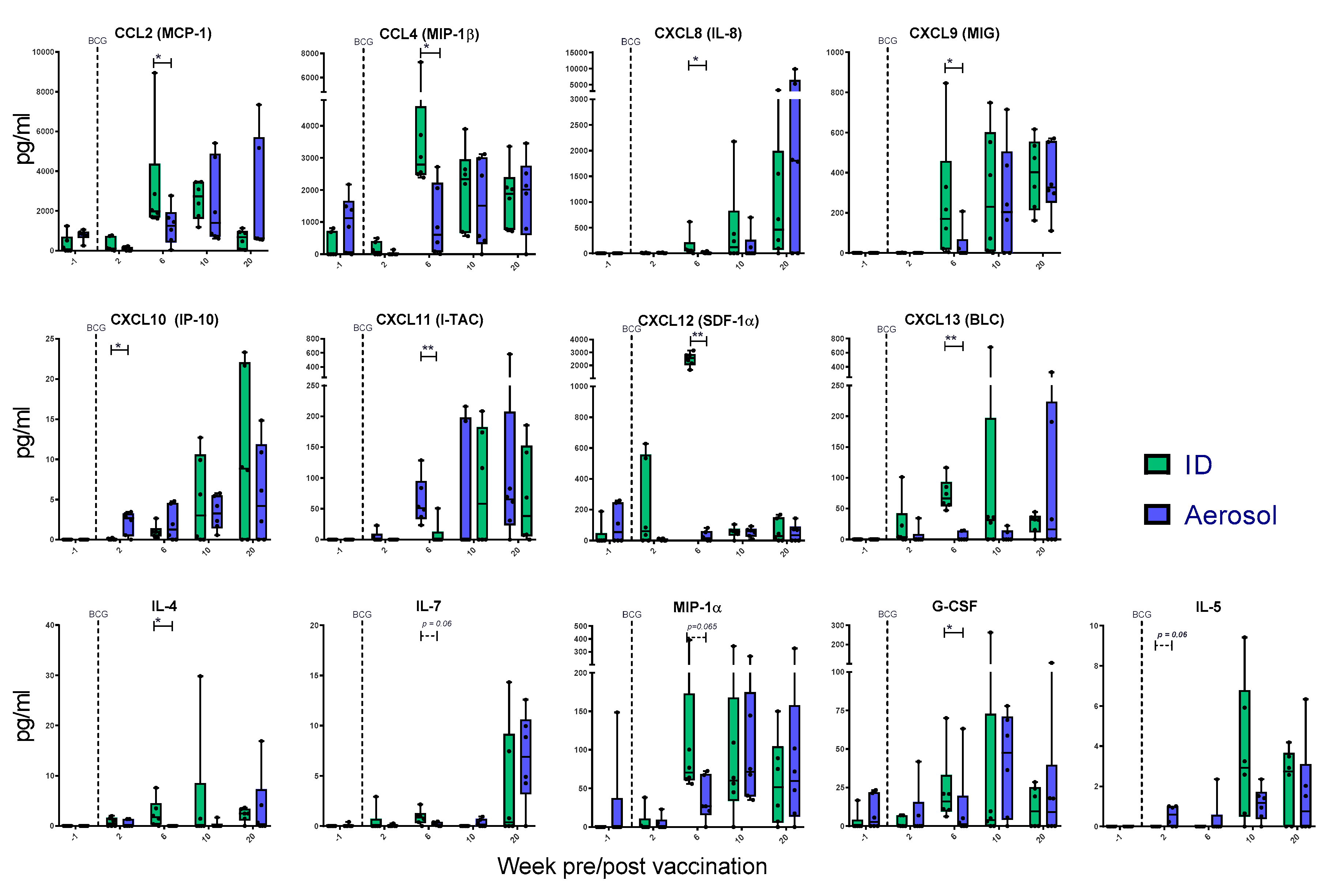
3.8. Mycobacterial Antigen-Specific Secretion of Biomarkers Measured by Cytokine Bead Array
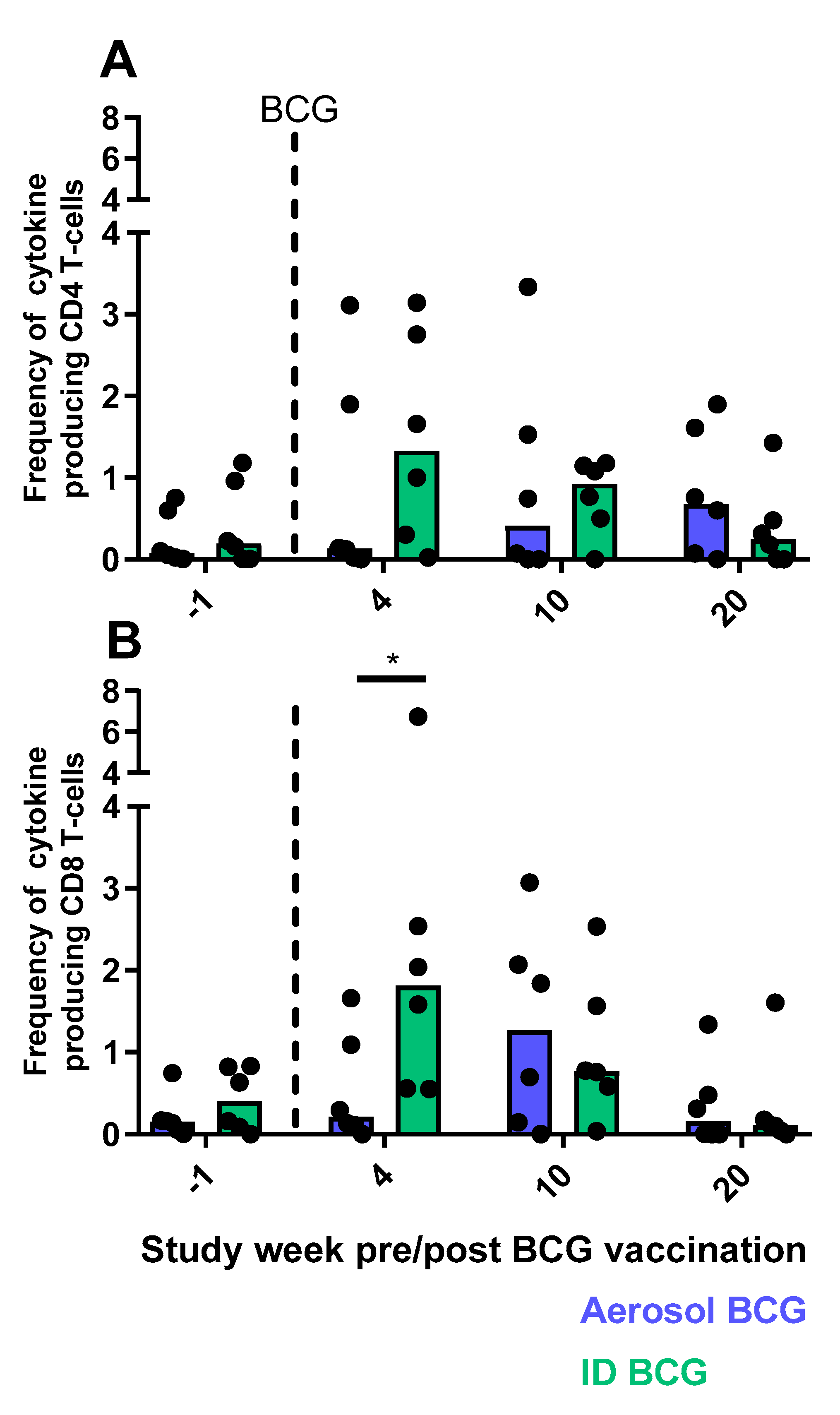
3.9. Cellular Immune Responses Measured by Flow Cytometry
3.9.1. CD4 and CD8 T-cell Functional Profiles Following Aerosol or ID BCG Vaccination
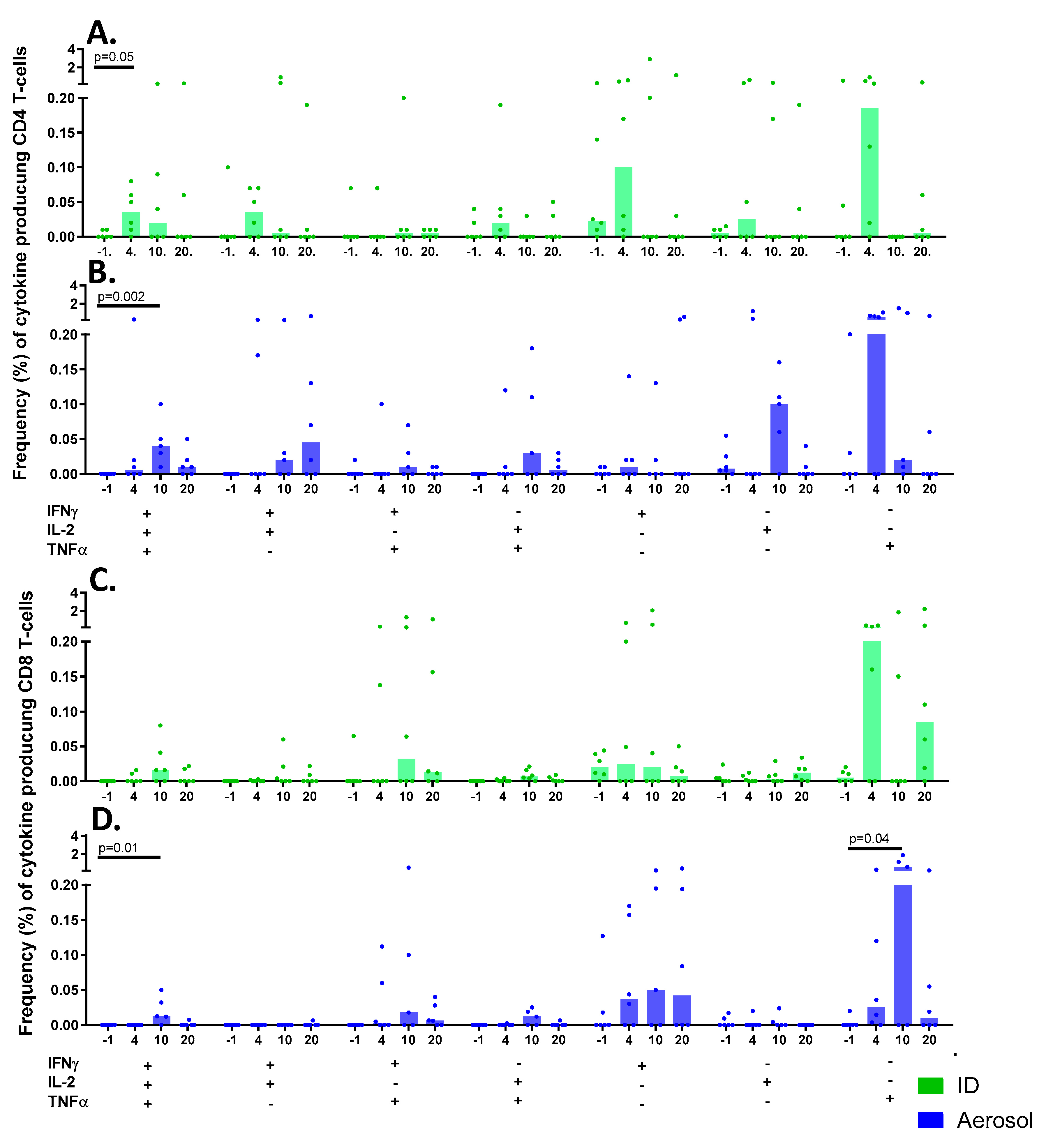
3.9.2. Multifunctional T-cell Populations Induced by Aerosol BCG or ID BCG Vaccination
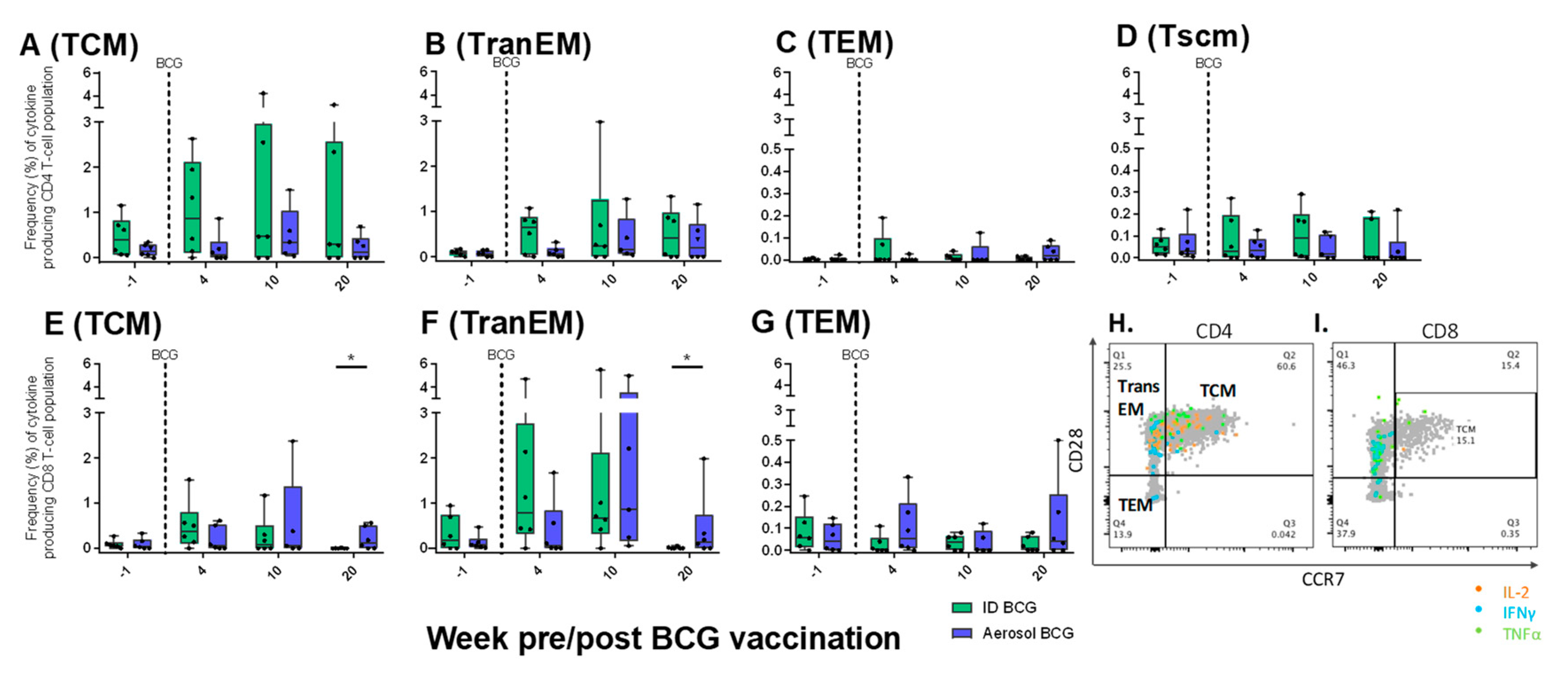
3.9.3. Antigen-Specific Memory T-cell Profiles Following Aerosol or ID BCG Vaccination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Capuano, S.V.; Croix, D.A.; Pawar, S.; Zinovik, A.; Myers, A.; Lin, P.L.; Bissel, S.; Fuhrman, C.; Klein, E.; Flynn, J.L. Experimental mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 2003, 71, 5831–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, S.A.; McShane, H.; Dennis, M.J.; Basaraba, R.J.; Gleeson, F.; Hall, G.; McIntyre, A.; Gooch, K.; Clark, S.; Beveridge, N.E.R.; et al. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clin. Vaccine Immunol. 2010, 17, 1170–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laddy, D.J.; Bonavia, A.; Hanekom, W.A.; Kaushal, D.; Williams, A.; Roederer, M.; Seder, R.A.; Sharpe, S.; Verreck, F.A.W.; Darrah, P.A. Toward tuberculosis vaccine development: Recommendations for nonhuman primate study design. Infect. Immun. 2018, 86, e00776-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, J.C.; Ho, W.-Z. Monkey models of tuberculosis: Lessons learned. Infect. Immun. 2015, 83, 852–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, S.; White, A.; Gleeson, F.; McIntyre, A.; Smyth, D.; Clark, S.; Sarfas, C.; Laddy, D.; Rayner, E.; Hall, G.; et al. Ultra low dose aerosol challenge with Mycobacterium tuberculosis leads to divergent outcomes in rhesus and cynomolgus macaques. Tuberculosis 2016, 96, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Trunz, B.B.; Fine, P.; Dye, C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: A meta-analysis and assessment of cost-effectiveness. Lancet 2006, 367, 1173–1180. [Google Scholar] [CrossRef]
- Colditz, G.A.; Brewer, T.F.; Berkey, C.S.; Wilson, M.E.; Burdick, E.; Fineberg, H.V.; Mosteller, F. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 1994, 271, 698–702. [Google Scholar] [CrossRef]
- Sterne, J.A.; Rodrigues, L.C.; Guedes, I.N. Does the efficacy of BCG decline with time since vaccination? Int. J. Tuberc. Lung Dis. 1998, 2, 200–207. [Google Scholar]
- Suliman, S.; Geldenhuys, H.; Johnson, J.L.; Hughes, J.E.; Smit, E.; Murphy, M.; Toefy, A.; Lerumo, L.; Hopley, C.; Pienaar, B.; et al. Bacillus calmette–guérin (BCG) revaccination of adults with latent Mycobacterium tuberculosis infection induces long-lived BCG-Reactive NK cell responses. J. Immunol. 2016, 197, 1100–1110. [Google Scholar] [CrossRef] [Green Version]
- Nemes, E.; Geldenhuys, H.; Rozot, V.; Tucker Rutkowski, K.; Ratangee, F.; Bilek, N.; Mabwe, S.; Makhethe, L.; Erasmus, M.; Toefy, A.; et al. Prevention of infection with Mycobacterium tuberculosis by H4:IC31® vaccination or BCG revaccination in adolescents. N. Engl. J. Med. 2018, 379, 138–149. [Google Scholar] [CrossRef]
- Stylianou, E.; Paul, M.J.; Reljic, R.; McShane, H. Mucosal delivery of tuberculosis vaccines: A review of current approaches and challenges. Expert Rev. Vaccines 2019, 18, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Nuermberger, E.L.; Yoshimatsu, T.; Tyagi, S.; Bishai, W.R.; Grosset, J.H. Paucibacillary tuberculosis in mice after prior aerosol immunization with Mycobacterium bovis BCG. Infect. Immun. 2004, 72, 1065–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheorghiu, M. BCG-induced mucosal immune responses. Int. J. Immunopharmacol. 1994, 16, 435–444. [Google Scholar] [CrossRef]
- Garcia-Contreras, L.; Wong, Y.-L.; Muttil, P.; Padilla, D.; Sadoff, J.; Derousse, J.; Germishuizen, W.A.; Goonesekera, S.; Elbert, K.; Bloom, B.R.; et al. Immunization by a bacterial aerosol. Proc. Natl. Acad. Sci. USA 2008, 105, 4656–4660. [Google Scholar] [CrossRef] [Green Version]
- Barclay, W.R.; Busey, W.M.; Dalgard, D.W.; Good, R.C.; Janicki, B.W.; Kasik, J.E.; Ribi, E.; Ulrich, C.E.; Wolinsky, E. Protection of monkeys against airborne tuberculosis by aerosol vaccination with bacillus Calmette-Guerin. Am. Rev. Respir. Dis. 1973, 107, 351–358. [Google Scholar]
- Zhou, Q.; Tang, P.; Leung, S.S.Y.; Chan, J.G.Y.; Chan, H.-K. Emerging inhalation aerosol devices and strategies: Where are we headed? Adv. Drug Deliv. Rev. 2014, 75, 3–17. [Google Scholar] [CrossRef]
- Waldrep, J.C.; Dhand, R. Advanced nebulizer designs employing vibrating mesh/aperture plate technologies for aerosol generation. Curr. Drug Deliv. 2008, 5, 114–119. [Google Scholar] [CrossRef]
- White, A.D.; Sarfas, C.; West, K.; Sibley, L.S.; Wareham, A.S.; Clark, S.; Dennis, M.J.; Williams, A.; Marsh, P.D.; Sharpe, S.A. An evaluation of the immunogenicity of BCG, delivered by aerosol to the lungs of macaques. Clin. Vaccine Immunol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, S.A.; Eschelbach, E.; Basaraba, R.J.; Gleeson, F.; Hall, G.A.; McIntyre, A.; Williams, A.; Kraft, S.L.; Clark, S.; Gooch, K.; et al. Determination of lesion volume by MRI and stereology in a macaque model of tuberculosis. Tuberculosis 2009, 89, 405–416. [Google Scholar] [CrossRef]
- Clark, S.; Hall, Y.; Kelly, D.; Hatch, G.; Williams, A. Survival of Mycobacterium tuberculosis during experimental aerosolization and implications for aerosol challenge models. J. Appl. Microbiol. 2011, 111, 350–359. [Google Scholar] [CrossRef]
- Druett, H.A. A mobile form of the Henderson apparatus. J. Hyg. 1969, 67, 437–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartings, J.M.; Roy, C.J. The automated bioaerosol exposure system: Preclinical platform development and a respiratory dosimetry application with nonhuman primates. J. Pharmacol. Toxicol. Methods 2004, 49, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, S.; White, A.; Sarfas, C.; Sibley, L.; Gleeson, F.; McIntyre, A.; Basaraba, R.; Clark, S.; Hall, G.; Rayner, E.; et al. Alternative BCG delivery strategies improve protection against Mycobacterium tuberculosis in non-human primates: Protection associated with mycobacterial antigen-specific CD4 effector memory T-cell populations. Tuberculosis 2016, 101, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, E.L.; Pearson, G.R.; Hall, G.A.; Gleeson, F.; McIntyre, A.; Smyth, D.; Dennis, M.J.; Sharpe, S.A. Early lesions following aerosol challenge of rhesus macaques (Macaca mulatta) with Mycobacterium tuberculosis (Erdman strain). J. Comp. Pathol. 2015, 152, 217–226. [Google Scholar] [CrossRef]
- Dennis, M.J.; Parks, S.; Bell, G.; Taylor, I.; Lakeman, J.; Sharpe, S.A. A flexible approach to imaging in ABSL-3 laboratories. Appl. Biosaf. 2015, 20, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Black, G.F.; Fine, P.N.; Warndorff, D.K.; Floyd, S.; Weir, R.E.; Blackwell, J.M.; Bliss, L.; Sichali, L.; Mwaungulu, L.; Chaguluka, S.; et al. Relationship between IFN-gamma and skin test responsiveness to Mycobacterium tuberculosis PPD in healthy, non-BCG-vaccinated young adults in Northern Malawi. Int. J. Tuberc. Lung Dis. 2001, 5, 664–672. [Google Scholar]
- Rayner, E.L.; Pearson, G.R.; Hall, G.A.; Basaraba, R.J.; Gleeson, F.; McIntyre, A.; Clark, S.; Williams, A.; Dennis, M.J.; Sharpe, S.A. Early lesions following aerosol infection of rhesus macaques (Macaca mulatta) with Mycobacterium tuberculosis strain H37RV. J. Comp. Pathol. 2013, 149, 475–485. [Google Scholar] [CrossRef]
- Roederer, M.; Nozzi, J.L.; Nason, M.C. SPICE: Exploration and analysis of post-cytometric complex multivariate datasets. Cytom. A 2011, 79, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Min, F.; Wu, R.; Pan, J.; Huang, S.; Luo, Y.; Zhang, Y. Positive correlation between IP-10 and IFN-γ levels in rhesus monkeys (Macaca mulatta) with either naturally acquired or experimental infection of Mycobacterium tuberculosis. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Ruhwald, M.; Aabye, M.G.; Ravn, P. IP-10 release assays in the diagnosis of tuberculosis infection: Current status and future directions. Expert Rev. Mol. Diagn. 2012, 12, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Takatsu, K. Interleukin-5 and IL-5 receptor in health and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 463–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diedrich, C.R.; Mattila, J.T.; Flynn, J.L. Monocyte-Derived IL-5 Reduces TNF Production by Mycobacterium tuberculosis—Specific CD4 T cells during SIV/M. tuberculosis coinfection. J. Immunol. 2013, 190, 6320–6328. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, C.J.; Hagen, S.I.; Walker, J.M.; Lum, R.; Mitchell, B.L.; Maino, V.C.; Axthelm, M.K.; Picker, L.J. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 2002, 168, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Picker, L.J.; Reed-Inderbitzin, E.F.; Hagen, S.I.; Edgar, J.B.; Hansen, S.G.; Legasse, A.; Planer, S.; Piatak, M.; Lifson, J.D.; Maino, V.C.; et al. IL-15 induces CD4+ effector memory T cell production and tissue emigration in nonhuman primates. J. Clin. Invest. 2006, 116, 1514–1524. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- WHO Global Tuberculosis Report 2018. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 25 October 2018).
- Sharpe, S.A.; White, A.D.; Sibley, L.; Gleeson, F.; Hall, G.A.; Basaraba, R.J.; McIntyre, A.; Clark, S.O.; Gooch, K.; Marsh, P.D.; et al. An aerosol challenge model of tuberculosis in Mauritian cynomolgus macaques. PLoS ONE 2017, 12, e0171906. [Google Scholar] [CrossRef]
- Rahman, S.; Magalhaes, I.; Rahman, J.; Ahmed, R.K.; Sizemore, D.R.; Scanga, C.A.; Weichold, F.; Verreck, F.; Kondova, I.; Sadoff, J.; et al. Prime-boost vaccination with rBCG/rAd35 enhances CD8+ cytolytic T-cell responses in lesions from Mycobacterium tuberculosis—Infected primates. Mol. Med. 2012, 18, 647–658. [Google Scholar] [CrossRef]
- Verreck, F.A.W.; Tchilian, E.Z.; Vervenne, R.A.W.; Sombroek, C.C.; Kondova, I.; Eissen, O.A.; Sommandas, V.; van der Werff, N.M.; Verschoor, E.; Braskamp, G.; et al. Variable BCG efficacy in rhesus populations: Pulmonary BCG provides protection where standard intra-dermal vaccination fails. Tuberculosis 2017, 104, 46–57. [Google Scholar] [CrossRef]
- Langermans, J.A.M.; Andersen, P.; van Soolingen, D.; Vervenne, R.A.W.; Frost, P.A.; van der Laan, T.; van Pinxteren, L.A.H.; van den Hombergh, J.; Kroon, S.; Peekel, I.; et al. Divergent effect of bacillus Calmette–Guérin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: Implications for primate models in tuberculosis vaccine research. Proc. Natl. Acad. Sci. USA 2001, 98, 11497–11502. [Google Scholar] [CrossRef] [Green Version]
- Darrah, P.A.; Zeppa, J.J.; Maiello, P.; Hackney, J.A.; Wadsworth, M.H.; Hughes, T.K.; Pokkali, S.; Swanson, P.A.; Grant, N.L.; Rodgers, M.A.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Sagalla, R.B.; Smaldone, G.C. Capturing the efficiency of vibrating mesh nebulizers: Minimizing upper airway deposition. J. Aerosol. Med. Pulm. Drug Deliv. 2014, 27, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Aerosol BCG Challenge Trial in Healthy UK Adults—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02709278 (accessed on 13 December 2019).
- Investigating Immune Responses to Aerosol BCG Challenge in Healthy UK Adults—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03912207 (accessed on 13 December 2019).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Vaccine and Route | Animal Identification Number | Presented Dose (cfu) | Estimated Retained Dose (cfu) |
|---|---|---|---|---|
| Group A | BCG Aerosol | U12 | 23 | 3 |
| U26 | 22 | 3 | ||
| U41 | 23 | 3 | ||
| U46 | 25 | 4 | ||
| U58 | 23 | 3 | ||
| U66 | 25 | 4 | ||
| Group B | BCG ID | U18 | 22 | 3 |
| U28 | 27 | 4 | ||
| U31 | 21 | 3 | ||
| U56 | 24 | 3 | ||
| U72 | 23 | 3 | ||
| Group C | No vaccine | U15 | 19 | 3 |
| U22 | 18 | 3 | ||
| U36 | 23 | 3 | ||
| U49 | 23 | 3 | ||
| U57 | 26 | 4 | ||
| U60 | 23 | 3 | ||
| U73 | 21 | 3 | ||
| Study median | 23 | 3 | ||
| Group | Vaccine and Route | Animal ID | Pulmonary | Extra-Pulmonary Tissues | Lymph Nodes | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lung Lesion Type (1–6) | Bronchus Associated Lymphoid Tissue Involvement | Multi-Nucleated Giant Cells | Spleen | Liver | Kidney | Lung Associated | Peripheral | |||
| (n of 7 Lung Lobes) | Present (+) or Absent (-) | |||||||||
| Group A | BCG Aerosol | U12 | 1, 2, 4, 5 | 0 | 5 | - | - | - | + | - |
| U26 | 1, 2, 4, 5, 6 | 0 | 6 | + | + | - | + | - | ||
| U41 | 1, 2, 4, 5, 6 | 0 | 7 | + | + | + | + | - | ||
| U46 | 1, 4, 5, 6 | 1 | 7 | - | - | - | + | - | ||
| U58 | 1, 2, 4, 5 | 3 | 7 | - | - | - | + | - | ||
| U66 | 2, 3, 4, 5, 6 | 0 | 6 | - | + | + | + | - | ||
| Group B | BCG ID | U18 | 1, 2, 4, 5, 6 | 1 | 5 | - | + | + | + | + |
| U28 | 1, 2, 3, 4, 5, 6 | 3 | 7 | + | + | + | + | - | ||
| U31 | 1, 2, 4, 5, 6 | 1 | 6 | + | + | + | + | + | ||
| U56 | 1, 4, 5, 6 | 0 | 3 | + | + | - | + | + | ||
| U72 | 1, 4, 5, 6 | 0 | 4 | + | + | - | + | - | ||
| Group C | No vaccine | U15 | 1, 4, 5, 6 | 0 | 5 | + | + | + | + | + |
| U22 | 1, 2, 4, 5, 6 | 0 | 6 | + | + | + | + | + | ||
| U36 | 1, 2, 4, 5, 6 | 1 | 6 | + | + | + | + | - | ||
| U49 | 1, 2, 3, 4, 5, 6 | 0 | 7 | + | + | + | + | - | ||
| U57 | 4, 5, 6 | 0 | 4 | - | - | + | + | + | ||
| U60 | 1, 2, 4, 5, 6 | 0 | 5 | + | + | + | + | + | ||
| U73 | 1, 4, 5, 6 | 0 | 7 | + | + | + | + | + | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, A.D.; Sarfas, C.; Sibley, L.S.; Gullick, J.; Clark, S.; Rayner, E.; Gleeson, F.; Català, M.; Nogueira, I.; Cardona, P.-J.; et al. Protective Efficacy of Inhaled BCG Vaccination Against Ultra-Low Dose Aerosol M. tuberculosis Challenge in Rhesus Macaques. Pharmaceutics 2020, 12, 394. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12050394
White AD, Sarfas C, Sibley LS, Gullick J, Clark S, Rayner E, Gleeson F, Català M, Nogueira I, Cardona P-J, et al. Protective Efficacy of Inhaled BCG Vaccination Against Ultra-Low Dose Aerosol M. tuberculosis Challenge in Rhesus Macaques. Pharmaceutics. 2020; 12(5):394. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12050394
Chicago/Turabian StyleWhite, Andrew D., Charlotte Sarfas, Laura S. Sibley, Jennie Gullick, Simon Clark, Emma Rayner, Fergus Gleeson, Martí Català, Isabel Nogueira, Pere-Joan Cardona, and et al. 2020. "Protective Efficacy of Inhaled BCG Vaccination Against Ultra-Low Dose Aerosol M. tuberculosis Challenge in Rhesus Macaques" Pharmaceutics 12, no. 5: 394. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12050394