A Physiologically-Based Pharmacokinetic Model of Trimethoprim for MATE1, OCT1, OCT2, and CYP2C8 Drug–Drug–Gene Interaction Predictions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
2.2. Trimethoprim Clinical Data
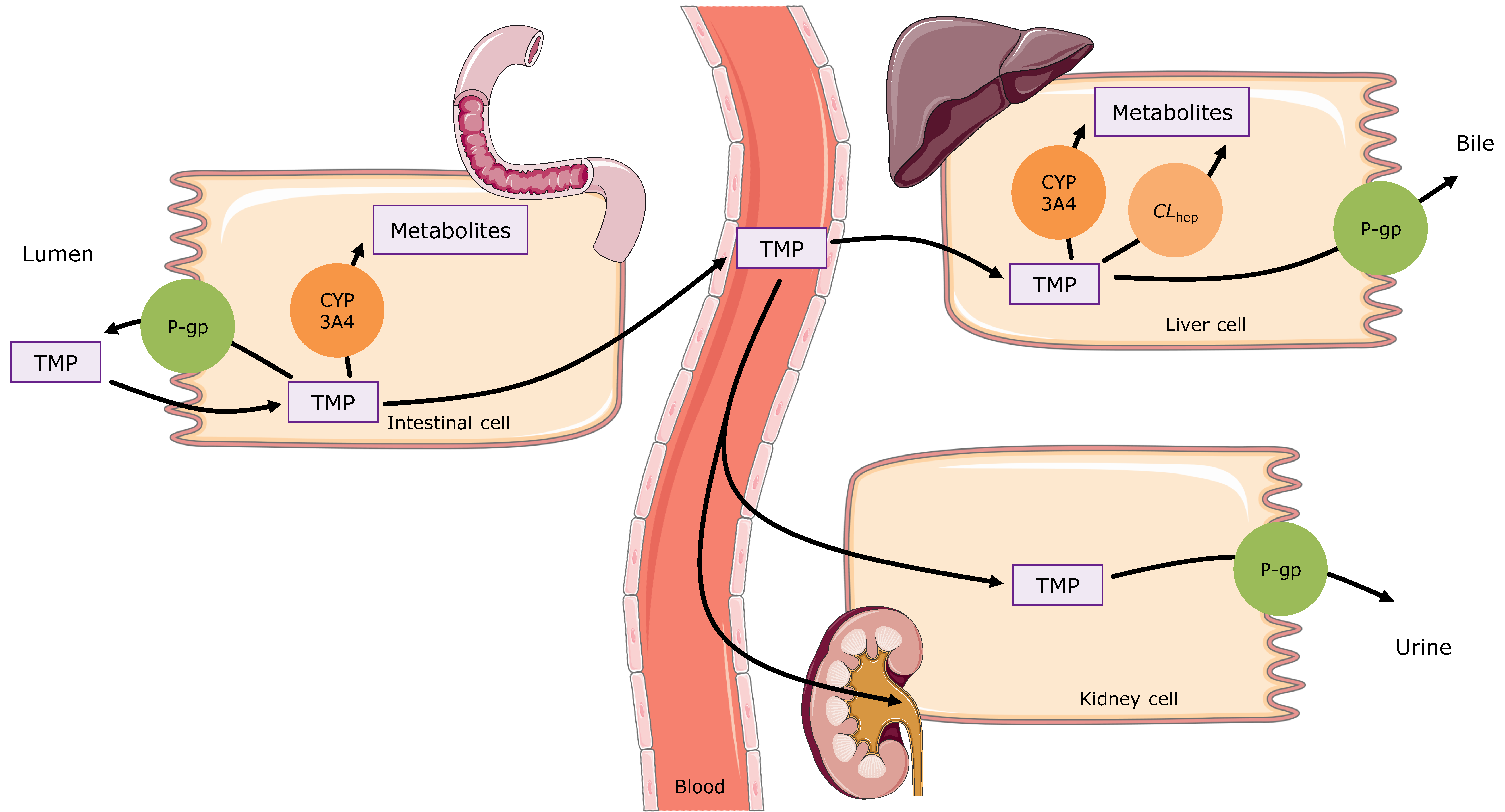
2.3. Trimethoprim PBPK Model Building
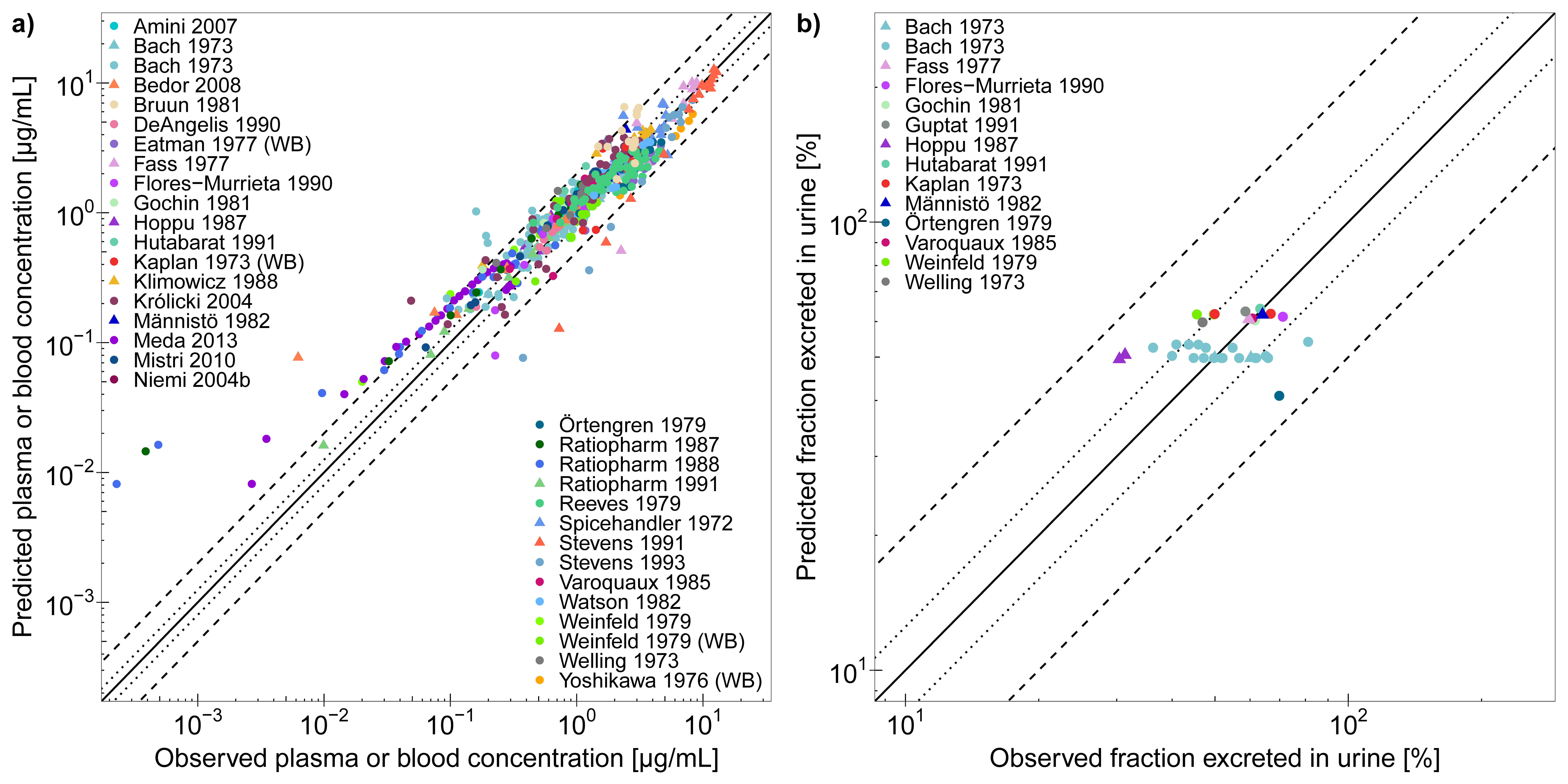
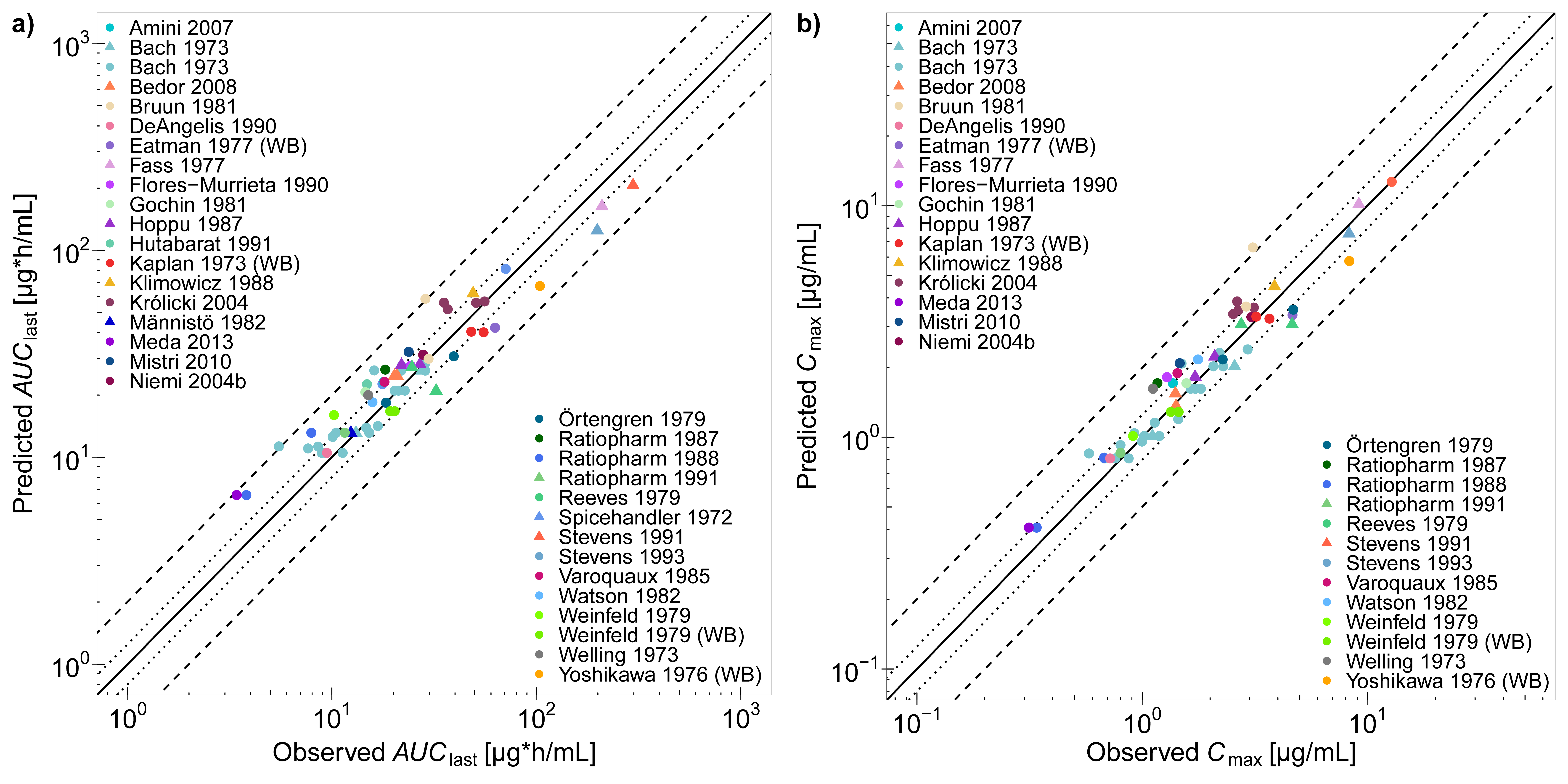
2.4. Trimethoprim PBPK Model Evaluation
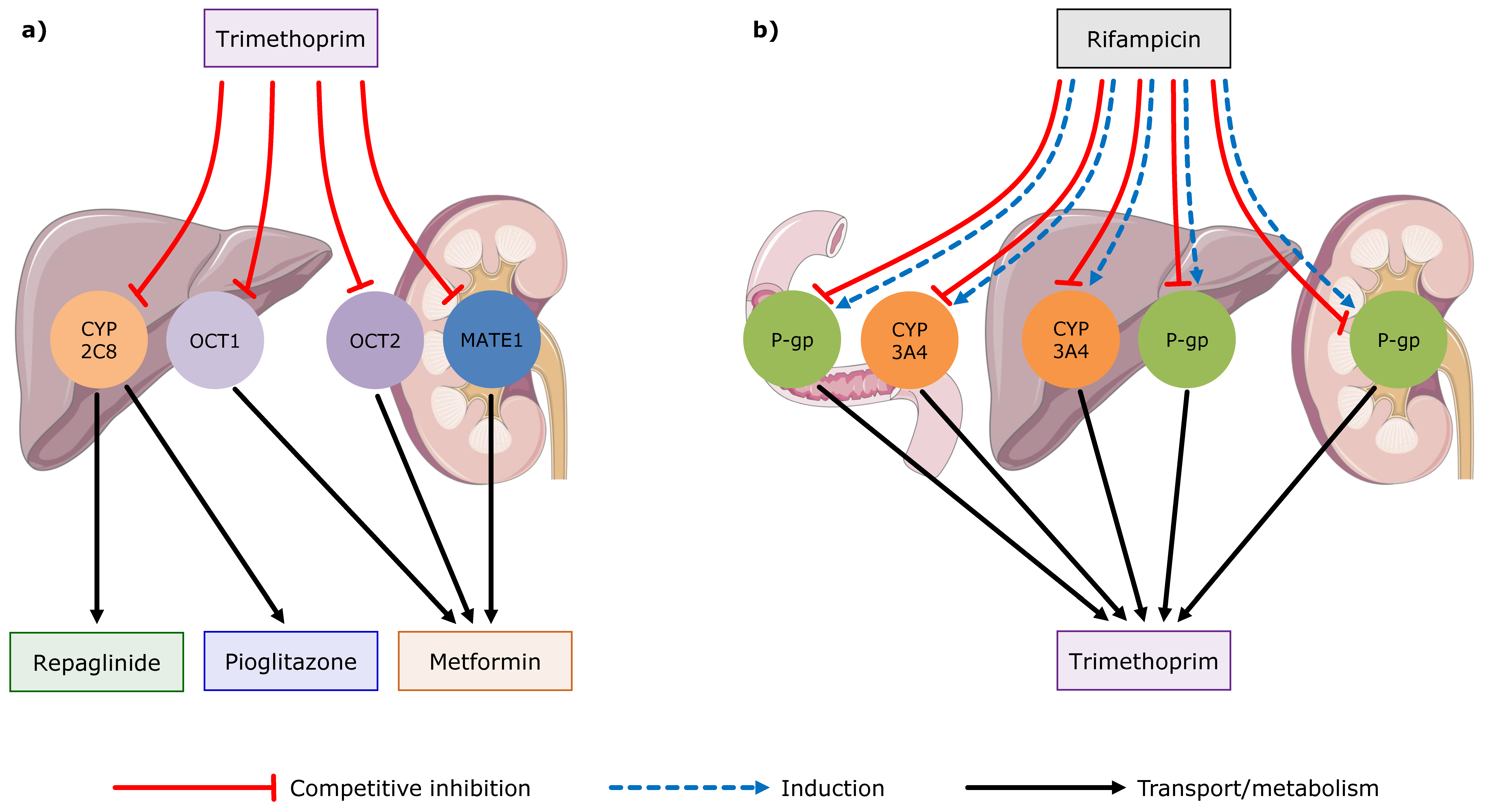
2.5. DDI and DDGI Modeling
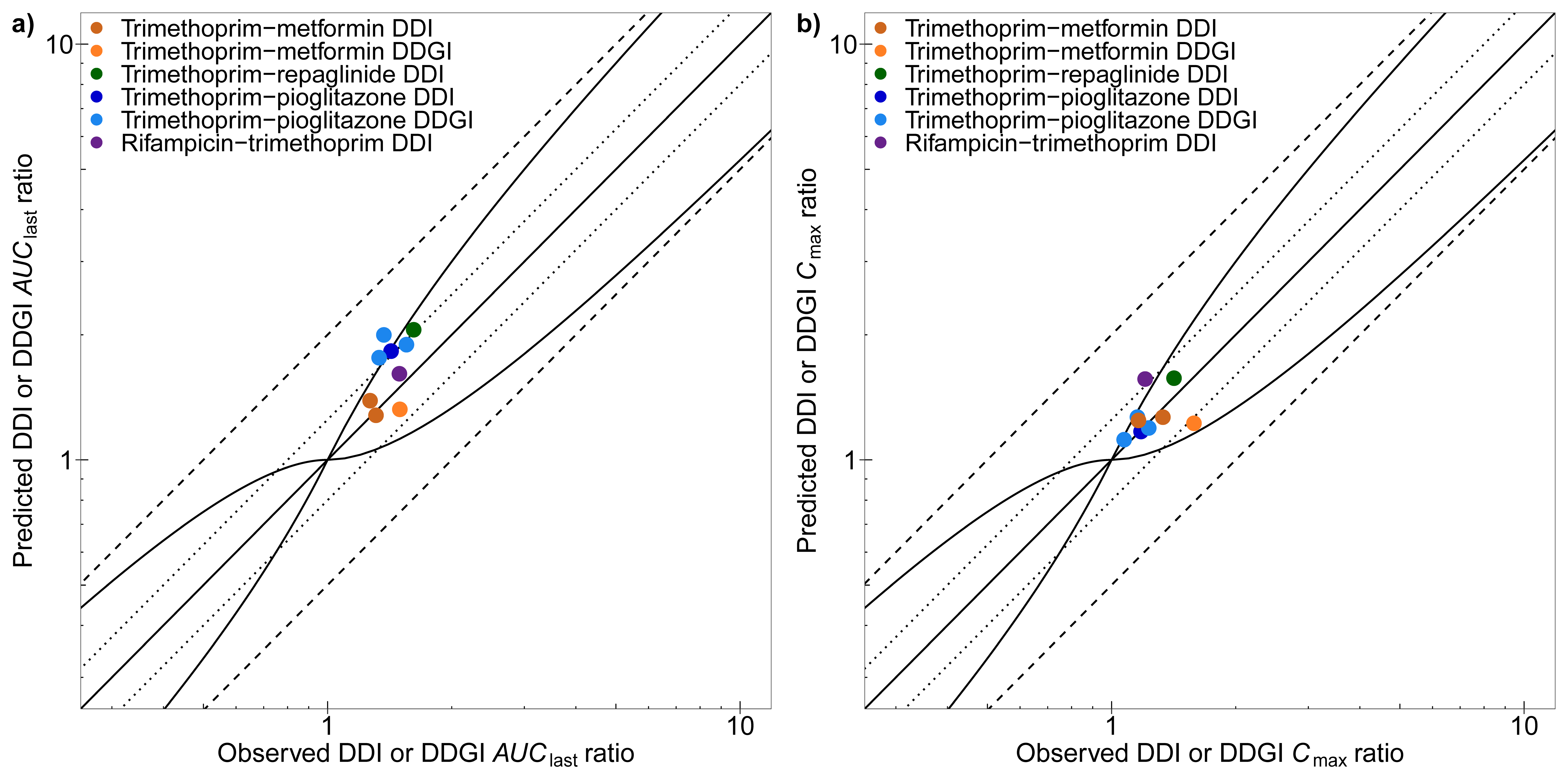
2.6. DDI and DDGI Model Performance Evaluation
2.7. Sensitivity Analysis
3. Results
3.1. Trimethoprim PBPK Model Building and Evaluation
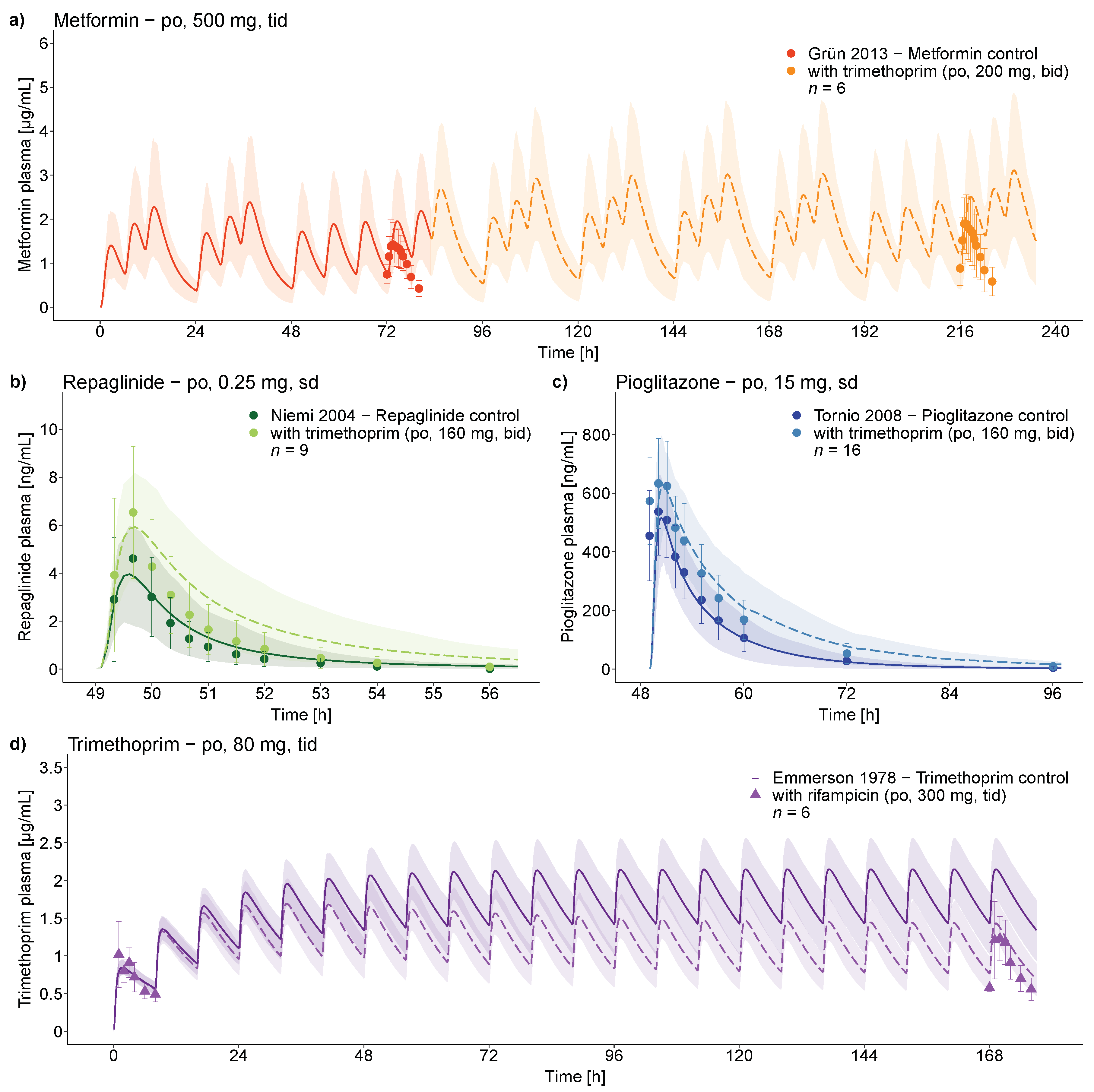
3.2. Trimethoprim DDI and DDGI Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Van Boeckel, T.P.; Gandra, S.; Ashok, A.; Caudron, Q.; Grenfell, B.T.; Levin, S.A.; Laxminarayan, R. Global antibiotic consumption 2000 to 2010: An analysis of national pharmaceutical sales data. Lancet. Infect. Dis. 2014, 14, 742–750. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 24 August 2020).
- Elsby, R.; Chidlaw, S.; Outteridge, S.; Pickering, S.; Radcliffe, A.; Sullivan, R.; Jones, H.; Butler, P. Mechanistic in vitro studies confirm that inhibition of the renal apical efflux transporter multidrug and toxin extrusion (MATE) 1, and not altered absorption, underlies the increased metformin exposure observed in clinical interactions with cimetidine, trimethoprim or pyrimethamine. Pharmacol. Res. Perspect. 2017, 5, 1–13. [Google Scholar]
- Müller, F.; Pontones, C.A.; Renner, B.; Mieth, M.; Hoier, E.; Auge, D.; Maas, R.; Zolk, O.; Fromm, M.F. N(1)-methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: Studies on the metformin-trimethoprim interaction. Eur. J. Clin. Pharmacol. 2015, 71, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Kajosaari, L.I.; Neuvonen, M.; Backman, J.T.; Neuvonen, P.J. The CYP2C8 inhibitor trimethoprim increases the plasma concentrations of repaglinide in healthy subjects. Br. J. Clin. Pharmacol. 2004, 57, 441–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornio, A.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab. Dispos. 2008, 36, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grün, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.-D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprim-metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharmacol. 2013, 76, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information (NCBI) dbSNP–rs316019. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/snp/rs316019 (accessed on 16 October 2020).
- Wang, Z.-J.; Yin, O.Q.P.; Tomlinson, B.; Chow, M.S.S. OCT2 polymorphisms and in-vivo renal functional consequence: Studies with metformin and cimetidine. Pharmacogenet. Genom. 2008, 18, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, S.; Brown, C.; Cheatham, S.; Castro, R.A.; Leabman, M.K.; Urban, T.J.; Chen, L.; Yee, S.W.; Choi, J.H.; et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet. Genom. 2009, 19, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, M.M.H.; Pedersen, R.S.; Stage, T.B.; Brasch-Andersen, C.; Nielsen, F.; Damkier, P.; Beck-Nielsen, H.; Brøsen, K. A gene-gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet. Genom. 2013, 23, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Clemens, E.; Broer, L.; Langer, T.; Uitterlinden, A.G.; de Vries, A.C.H.; van Grotel, M.; Pluijm, S.F.M.; Binder, H.; Byrne, J.; van Dulmen-den Broeder, E.; et al. Genetic variation of cisplatin-induced ototoxicity in non-cranial-irradiated pediatric patients using a candidate gene approach: The International PanCareLIFE Study. Pharm. J. 2020, 20, 294–305. [Google Scholar] [CrossRef]
- Dai, D.; Zeldin, D.C.; Blaisdell, J.A.; Chanas, B.; Coulter, S.J.; Ghanayem, B.I.; Goldstein, J.A. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 2001, 11, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Leathart, J.B.; Neuvonen, M.; Backman, J.T.; Daly, A.K.; Neuvonen, P.J. Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin. Pharmacol. Ther. 2003, 74, 380–387. [Google Scholar] [CrossRef]
- Greiner, B.; Eichelbaum, M.; Fritz, P.; Kreichgauer, H.P.; von Richter, O.; Zundler, J.; Kroemer, H.K. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 1999, 104, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmerson, A.M.; Grüneberg, R.N.; Johnson, E.S. The phamacokinetics in man of a combination of rifampicin and trimethoprim. J. Antimicrob. Chemother. 1978, 4, 523–531. [Google Scholar] [CrossRef]
- Mitchell, M.; Muftakhidinov, B.; Winchen, T.; Jędrzejewski-Szmek, Z.; Trande, A.; Weingrill, J.; Langer, S.; Lane, D.; Sower, K. Engauge Digitizer Software. Available online: https://markummitchell.github.io/engauge-digitizer (accessed on 24 August 2020).
- Wojtyniak, J.-G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data aigitizing: Accurate and precise data extraction for quantitative systems pharmacology and physiologically-based pharmacokinetic modeling. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 322–331. [Google Scholar] [CrossRef]
- Open Systems Pharmacology Suite Community. PK-Sim® Ontogeny Database Documentation, Version 7.3. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim Ontogeny Database Version 7.3.pdf (accessed on 24 August 2020).
- Hanke, N.; Türk, D.; Selzer, D.; Ishiguro, N.; Ebner, T.; Wiebe, S.; Müller, F.; Stopfer, P.; Nock, V.; Lehr, T. A comprehensive whole-body physiologically based pharmacokinetic drug-drug-gene interaction model of metformin and cimetidine in healthy adults and renally impaired individuals. Clin. Pharmacokinet. 2020. [Google Scholar] [CrossRef]
- Türk, D.; Hanke, N.; Wolf, S.; Frechen, S.; Eissing, T.; Wendl, T.; Schwab, M.; Lehr, T. Physiologically based pharmacokinetic models for prediction of complex CYP2C8 and OATP1B1 (SLCO1B1) drug-drug-gene interactions: A modeling network of gemfibrozil, repaglinide, pioglitazone, rifampicin, clarithromycin and itraconazole. Clin. Pharmacokinet. 2019, 58, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK models for CYP3A4 and P-gp DDI prediction: A modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Lepist, E.-I.; Zhang, X.; Hao, J.; Huang, J.; Kosaka, A.; Birkus, G.; Murray, B.P.; Bannister, R.; Cihlar, T.; Huang, Y.; et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014, 86, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.; König, J.; Glaeser, H.; Schmidt, I.; Zolk, O.; Fromm, M.F.; Maas, R. Molecular mechanism of renal tubular secretion of the antimalarial drug chloroquine. Antimicrob. Agents Chemother. 2011, 55, 3091–3098. [Google Scholar] [CrossRef] [Green Version]
- Lechner, C.; Ishiguro, N.; Fukuhara, A.; Shimizu, H.; Ohtsu, N.; Takatani, M.; Nishiyama, K.; Washio, I.; Yamamura, N.; Kusuhara, H. Impact of experimental conditions on the evaluation of interactions between multidrug and toxin extrusion proteins and candidate drugs. Drug Metab. Dispos. 2016, 44, 1381–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, F.; König, J.; Hoier, E.; Mandery, K.; Fromm, M.F. Role of organic cation transporter OCT2 and multidrug and toxin extrusion proteins MATE1 and MATE2-K for transport and drug interactions of the antiviral lamivudine. Biochem. Pharmacol. 2013, 86, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Bleasby, K.; Chan, G.H.; Nunes, I.; Evers, R. The complexities of interpreting reversible elevated serum creatinine levels in drug development: Does a correlation with inhibition of renal transporters exist? Drug Metab. Dispos. 2016, 44, 1498–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinger, J.; Meyer, M.R.; Maurer, H.H. Development of an in vitro cytochrome P450 cocktail inhibition assay for assessing the inhibition risk of drugs of abuse. Toxicol. Lett. 2014, 230, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Nolte, H.; Büttner, H. Pharmacokinetics of trimethoprim and its combination with sulfamethoxazole in man after single and chronic oral administration. Chemotherapy 1973, 18, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, S.A.; Weinfeld, R.E.; Abruzzo, C.W.; McFaden, K.; Lewis Jack, M.; Weissman, L. Pharmacokinetic profile of trimethoprim-sulfamethoxazole in man. J. Infect. Dis. 1973, 128, S547–S555. [Google Scholar] [CrossRef]
- Weinfeld, R.E.; Macasieb, T.C. Determination of trimethoprim in biological fluids by high-performance liquid chromatography. J. Chromatogr. B 1979, 164, 73–84. [Google Scholar] [CrossRef]
- Guptat, R.L.; Kumar, R.; Singla, A.K. Enhanced dissolution and absorption of trimethoprim from coprecipitates with polyethylene glycols and polyvinylpyrrolidone. Drug Dev. Ind. Pharm. 1991, 17, 463–468. [Google Scholar] [CrossRef]
- Ratiopharm GmbH. Fachinformation Cotrim-ratiopharm® 480 mg Tabletten, Cotrim forte-ratiopharm® 960 mg Tabletten (Bioavailability Study 1987 and 1991); Ratiopharm GmbH.: Ulm, Germany, 2017. [Google Scholar]
- Les Laboratoires Servier. Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 24 August 2020).
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- O’Neil, M.J.; Heckelman, P.E.; Koch, C.B.; Roman, K.J.; Kenny, C.M.; D’Arecca, M.R. The Merck Index: An. Encyclopedia of Chemicals, Drugs, and Biologicals, 14th ed.; O’Neil, M.J., Heckelman, P.E., Koch, C.B., Roman, K.J., Eds.; Merc and Co. Inc.: Whitehouse Station, NJ, USA, 2006. [Google Scholar]
- Reeves, D.S.; Wilkinson, P.J. The pharmacokinetics of trimethoprim and trimethoprim/sulphonamide combinations, including penetration into body tissues. Infection 1979, 7 (Suppl. 4), S330–S341. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresta, M.; Furneri, P.M.; Mezzasalma, E.; Nicolosi, V.M.; Puglisi, G. Correlation of trimethoprim and brodimoprim physicochemical and lipid membrane interaction properties with their accumulation in human neutrophils. Antimicrob. Agents Chemother. 1996, 40, 2865–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Varoquaux, O.; Lajoie, D.; Gobert, C.; Cordonnier, P.; Ducreuzet, C.; Pays, M.; Advenier, C. Pharmacokinetics of the trimethoprim-sulphamethoxazole combination in the elderly. Br. J. Clin. Pharmacol. 1985, 20, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijkström, A.; Westerlund, D. Plasma protein binding of sulphadiazine, sulphamethoxazole and trimethoprim determined by ultrafiltration. J. Pharm. Biomed. Anal. 1983, 1, 293–299. [Google Scholar] [CrossRef]
- Männistö, P.T.; Mäntylä, R.; Mattila, J.; Nykänen, S.; Lamminsivu, U. Comparison of pharmacokinetics of sulphadiazine and sulphamethoxazole after intravenous infusion. J. Antimicrob. Chemother. 1982, 9, 461–470. [Google Scholar] [CrossRef]
- Hoffmann-La Roche Inc. BACTRIM TM Sulfamethoxazole and Trimethoprim DS (Double Strength) Tablets and Tablets USP; Hoffmann-La Roche Inc.: Basel, Switzerland, 2013. [Google Scholar]
- Singlas, E.; Colin, J.N.; Rottembourg, J.; Meessen, J.P.; de Martin, A.; Legrain, M.; Simon, P. Pharmacokinetics of sulfamethoxazole - trimethoprim combination during chronic peritoneal dialysis: Effect of peritonitis. Eur. J. Clin. Pharmacol. 1982, 21, 409–415. [Google Scholar] [CrossRef]
- Hutabarat, R.M.; Unadkat, J.D.; Sahajwalla, C.; McNamara, S.; Ramsey, B.; Smith, A.L. Disposition of drugs in cystic fibrosis. I. Sulfamethoxazole and trimethoprim. Clin. Pharmacol. Ther. 1991, 49, 402–409. [Google Scholar] [CrossRef]
- Gonzalez, D.; Schmidt, S.; Derendorf, H. Importance of relating efficacy measures to unbound drug concentrations for anti-infective agents. Clin. Microbiol. Rev. 2013, 26, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Berezhkovskiy, L.M. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J. Pharm. Sci. 2004, 93, 1628–1640. [Google Scholar] [CrossRef]
- Open Systems Pharmacology Suite Community. Open Systems Pharmacology Suite Manual, Version 7.4. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/Open Systems Pharmacology Suite.pdf (accessed on 24 August 2020).
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Bedor, D.C.G.; Gonçalves, T.M.; Ferreira, M.L.L.; de Sousa, C.E.M.; Menezes, A.L.; Oliveira, E.J.; de Santana, D.P. Simultaneous determination of sulfamethoxazole and trimethoprim in biological fluids for high-throughput analysis: Comparison of HPLC with ultraviolet and tandem mass spectrometric detection. J. Chromatogr. B 2008, 863, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ratiopharm GmbH. Fachinformation Cotrim K-ratiopharm® 240 mg/5 mL Saft, Cotrim E-ratiopharm® 480 mg / 5 mL Saft (Bioavailability Study 1988); Ratiopharm GmbH.: Ulm, Germany, 2013. [Google Scholar]
- Hoppu, K.; Tuomisto, J.; Koskimies, O.; Simell, O. Food and guar decrease absorption of trimethoprim. Eur. J. Clin. Pharmacol. 1987, 32, 427–429. [Google Scholar] [CrossRef]
- Bach, M.C.; Gold, O.; Finland, M. Absorption and urinary execretion of trimethoprim, sulfamethoxazole, and trimethoprim-sulfamethoxazole: Results with single doses in normal young adults and preliminary observations during therapy with trimethoprim-sulfamethoxazole. J. Infect. Dis. 1973, 128 (Suppl. 5), C84–C99. [Google Scholar] [CrossRef] [PubMed]
- Fass, R.J.; Prior, R.B.; Perkins, R.L. Pharmacokinetics and tolerance of a single twelve-tablet dose of trimethoprim (960 mg)-sulfamethoxazole (4800 mg). Antimicrob. Agents Chemother. 1977, 12, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Klimowicz, A.; Nowak, A.; Kadyków, M. Plasma and skin blister fluid concentrations of trimethoprim following its oral administration. Eur. J. Clin. Pharmacol. 1988, 34, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Laizure, S.C.; Williams, C.L.; Stein, D.S. Pharmacokinetics and adverse effects of 20-mg/kg/day trimethoprim and 100-mg/kg/day sulfamethoxazole in healthy adult subjects. Antimicrob. Agents Chemother. 1991, 35, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Spicehandler, J.; Pollock, A.A.; Simberkoff, M.S.; Rahal, J.J. Intravenous pharmacokinetics and in vitro bactericidal activity of trimethoprim-sulfamethoxazole. Rev. Infect. Dis. 1982, 4, 562–565. [Google Scholar] [CrossRef]
- Amini, H.; Ahmadiani, A. Rapid and simultaneous determination of sulfamethoxazole and trimethoprim in human plasma by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2007, 43, 1146–1150. [Google Scholar] [CrossRef]
- Bruun, J.N.; Ostby, N.; Bredesen, J.E.; Kierulf, P.; Lunde, P.K. Sulfonamide and trimethoprim concentrations in human serum and skin blister fluid. Antimicrob. Agents Chemother. 1981, 19, 82–85. [Google Scholar] [CrossRef] [Green Version]
- DeAngelis, D.V.; Woolley, J.L.; Sigel, C.W. High-performance liquid chromatographic assay for the simultaneous measurement of trimethoprim and sulfamethoxazole in plasma or urine. Ther. Drug Monit. 1990, 12, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Eatman, F.B.; Maggio, A.C.; Pocelinko, R.; Boxenbaum, H.G.; Geitner, K.A.; Glover, W.; Macasieb, T.; Holazo, A.; Weinfeld, R.E.; Kaplan, S.A. Blood and salivary concentrations of sulfamethoxazole and trimethoprim in man. J. Pharmacokinet. Biopharm. 1977, 5, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Flores-Murrieta, F.J.; Castañeda-Hernández, G.; Menéndez, J.C.; Chávez, F.; Herrera, J.E.; Hong, E. Pharmacokinetics of sulfamethoxazole and trimethoprim in Mexicans: Bioequivalence of two oral formulations (URO-TS D® and Bactrim F®). Biopharm. Drug Dispos. 1990, 11, 765–772. [Google Scholar] [CrossRef]
- Gochin, R.; Kanfer, I.; Haigh, J.M. Simultaneous determination of trimethoprim, sulphamethoxazole and N4-acetylsulphamethoxazole in serum and urine by high-performance liquid chromatography. J. Chromatogr. 1981, 223, 139–145. [Google Scholar] [CrossRef]
- Królicki, A.; Klimowicz, A.; Bielecka-Grzela, S.; Nowak, A.; Maleszka, R. Penetration of cotrimoxazole components into skin after a single oral dose. Theoretical versus experimental approach. Pol. J. Pharmacol. 2004, 56, 257–263. [Google Scholar]
- MEDA Pharma GmbH & Co KG. Fachinformation Cotrim-Diolan® Suspension Zum Einnehmen; MEDA Pharma GmbH & Co. KG: Bad Homburg, Germany, 2013. [Google Scholar]
- Mistri, H.N.; Jangid, A.G.; Pudage, A.; Shah, A.; Shrivastav, P.S. Simultaneous determination of sulfamethoxazole and trimethoprim in microgram quantities from low plasma volume by liquid chromatography–tandem mass spectrometry. Microchem. J. 2010, 94, 130–138. [Google Scholar] [CrossRef]
- Niemi, M.; Backman, J.T.; Neuvonen, P.J. Effects of trimethoprim and rifampin on the pharmacokinetics of the cytochrome P450 2C8 substrate rosiglitazone. Clin. Pharmacol. Ther. 2004, 76, 239–249. [Google Scholar] [CrossRef]
- Örtengren, B.; Magni, L.; Bergan, T. Development of sulphonamide-trimethoprim combinations for urinary tract infections. Part 3: Pharmacokinetic characterization of sulphadiazine and sulphamethoxazole given with trimethoprim. Infection 1979, 7, S371–S381. [Google Scholar] [CrossRef]
- Stevens, R.C.; Laizure, S.C.; Sanders, P.L.; Stein, D.S. Multiple-dose pharmacokinetics of 12 milligrams of trimethoprim and 60 milligrams of sulfamethoxazole per kilogram of body weight per day in healthy volunteers. Antimicrob. Agents Chemother. 1993, 37, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Watson, I.D.; Cohen, H.N.; Stewart, M.J.; McIntosh, S.J.; Shenkin, A.; Thomson, J.A. Comparative pharmacokinetics of co-trifamole and co-trimoxazole to “steady state” in normal subjects. Br. J. Clin. Pharmacol. 1982, 14, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Welling, P.G.; Craig, W.A.; Amidon, G.L.; Kunin, C.M. Pharmacokinetics of trimethoprim and sulfamethoxazole in normal subjects and in patients with renal failure. J. Infect. Dis. 1973, 128, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.T.; Guze, L.B. Concentrations of trimethoprim-sulfamethoxazole in blood after a single, large oral dose. Antimicrob. Agents Chemother. 1976, 10, 462–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the two-fold measure of prediction success for ratios: Application for the assessment of drug-drug interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susanto, M.; Benet, L.Z. Can the enhanced renal clearance of antibiotics in cystic fibrosis patients be explained by P-glycoprotein transport? Pharm. Res. 2002, 19, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Kito, T.; Ito, S.; Mizuno, T.; Maeda, K.; Kusuhara, H. Investigation of non-linear Mate1-mediated efflux of trimethoprim in the mouse kidney as the mechanism underlying drug-drug interactions between trimethoprim and organic cations in the kidney. Drug Metab. Pharmacokinet. 2019, 34, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Johnson, K.; Billington, S.; Lee, C.; Chung, G.W.; Brown, C.D.A.; Kelly, E.J.; Himmelfarb, J.; Unadkat, J.D. Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab. Dispos. 2016, 44, 1920–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribera, E.; Pou, L.; Fernandez-Sola, A.; Campos, F.; Lopez, R.M.; Ocaña, I.; Ruiz, I.; Pahissa, A. Rifampin reduces concentrations of trimethoprim and sulfamethoxazole in serum in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 2001, 45, 3238–3241. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Wang, J.; Backman, J.T.; Laitila, J.; Neuvonen, P.J. Trimethoprim and sulfamethoxazole are selective inhibitors of CYP2C8 and CYP2C9, respectively. Drug Metab. Dispos. 2002, 30, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Rylance, G.W.; George, R.H.; Healing, D.E.; Roberts, D.G. Single dose pharmacokinetics of trimethoprim. Arch. Dis. Child. 1985, 60, 29–33. [Google Scholar] [CrossRef] [Green Version]
- ClinCalc LLC. ClinCalc DrugStats Database. Available online: https://clincalc.com/DrugStats/ (accessed on 30 October 2020).
- Rowland Yeo, K.; Kenny, J.R.; Rostami-Hodjegan, A. Application of in vitro-in vivo extrapolation (IVIVE) and physiologically based pharmacokinetic (PBPK) modelling to investigate the impact of the CYP2C8 polymorphism on rosiglitazone exposure. Eur. J. Clin. Pharmacol. 2013, 69, 1311–1320. [Google Scholar]
- Scotcher, D.; Arya, V.; Yang, X.; Zhao, P.; Zhang, L.; Huang, S.-M.; Rostami-Hodjegan, A.; Galetin, A. A novel physiologically based model of creatinine renal disposition to integrate current knowledge of systems parameters and clinical observations. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Nakada, T.; Kudo, T.; Kume, T.; Kusuhara, H.; Ito, K. Quantitative analysis of elevation of serum creatinine via renal transporter inhibition by trimethoprim in healthy subjects using physiologically-based pharmacokinetic model. Drug Metab. Pharmacokinet. 2018, 33, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.J.; Wu, H.; Maharaj, A.; Edginton, A.N.; Balevic, S.J.; Cobbaert, M.; Cunningham, A.P.; Hornik, C.P.; Cohen-Wolkowiez, M. Physiologically based pharmacokinetic modeling for trimethoprim and sulfamethoxazole in children. Clin. Pharmacokinet. 2019, 58, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open Systems Pharmacology Community - An open access, open source, open science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 878–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Unit | Source | Literature | Reference | Description |
|---|---|---|---|---|---|---|
| MW | 290.32 | g/mol | Literature | 290.32 | [35] | Molecular weight |
| pKa (base) | 7.12 | - | Literature | 6.60, 7.12, 7.30 | [35,36,37] | Acid dissociation constant |
| Solubility (pH 7.0) | 0.40 | g/L | Literature | 0.40 | [35] | Solubility |
| logP | 1.01 | - | Optimized | 0.60, 0.73, 0.91, 1.43 | [35,38,39,40] | Lipophilicity |
| fu | 56 | % | Literature | 42–65 | [41,42,43,44,45,46,47] | Fraction unbound plasma |
| P-gp KM | 195.75 | µmol/L | Optimized | - | - | Michaelis –Menten constant |
| P-gp kcat | 1.44 | 1/min | Optimized | - | - | Transport rate constant |
| CYP3A4 KM | 375.57 | µmol/L | Optimized | - | - | Michaelis–Menten constant |
| CYP3A4 kcat | 0.56 | 1/min | Optimized | - | - | Catalytic rate constant |
| CLhep | 1.61 × 10−2 | 1/min | Optimized | - | - | Hepatic metabolic clearance |
| GFR fraction | 1 | - | Assumed | - | - | Fraction of filtered drug in the urine |
| EHC continuous fraction | 1 | - | Assumed | - | - | Fraction of bile continually released |
| MATE1 Ki | 4.45 | µmol/L | Literature | 0.51, 2.64, 3.29, 3.94, 4.06, 4.58, 6.30, 6.73, 7.99 * | [3,4,23,24,25] | Conc. for 50% inhibition (competitive) |
| OCT1 Ki | 32.20 | µmol/L | Literature | 27.70, 36.70 * | [3,4] | Conc. for 50% inhibition (competitive) |
| OCT2 Ki | 47.82 | µmol/L | Literature | 13.20, 19.80, 27.20, 32.30, 57.40, 137.00 * | [3,4,23,26,27] | Conc. for 50% inhibition (competitive) |
| CYP2C8 Ki | 4.85 | µmol/L | Literature | 2.25, 3.80, 8.50 * | [28] | Conc. for 50% inhibition (competitive) |
| Partition coefficients | Diverse | - | Calculated | Berezhkovskiy | [48] | Cell to plasma partition coefficients |
| Cellular permeability | 4.96 × 10−4 | cm/min | Calculated | CDS | [49] | Permeability into the cellular space |
| Intestinal permeability | 1.24 × 10−2 | cm/min | Optimized | 1.36 × 10−6 | Calculated | Transcellular intestinal permeability |
| Formulation | Weibull ° | - | Optimized | - | - | Formulation used in predictions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Türk, D.; Hanke, N.; Lehr, T. A Physiologically-Based Pharmacokinetic Model of Trimethoprim for MATE1, OCT1, OCT2, and CYP2C8 Drug–Drug–Gene Interaction Predictions. Pharmaceutics 2020, 12, 1074. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111074
Türk D, Hanke N, Lehr T. A Physiologically-Based Pharmacokinetic Model of Trimethoprim for MATE1, OCT1, OCT2, and CYP2C8 Drug–Drug–Gene Interaction Predictions. Pharmaceutics. 2020; 12(11):1074. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111074
Chicago/Turabian StyleTürk, Denise, Nina Hanke, and Thorsten Lehr. 2020. "A Physiologically-Based Pharmacokinetic Model of Trimethoprim for MATE1, OCT1, OCT2, and CYP2C8 Drug–Drug–Gene Interaction Predictions" Pharmaceutics 12, no. 11: 1074. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12111074