
Pharmacological Inhibition of STAT3 by Stattic Ameliorates Clinical Symptoms and Reduces Autoinflammation in Myeloid, Lymphoid, and Neuronal Tissue Compartments in Relapsing–Remitting Model of Experimental Autoimmune Encephalomyelitis in SJL/J Mice

 , , , and
, , , and 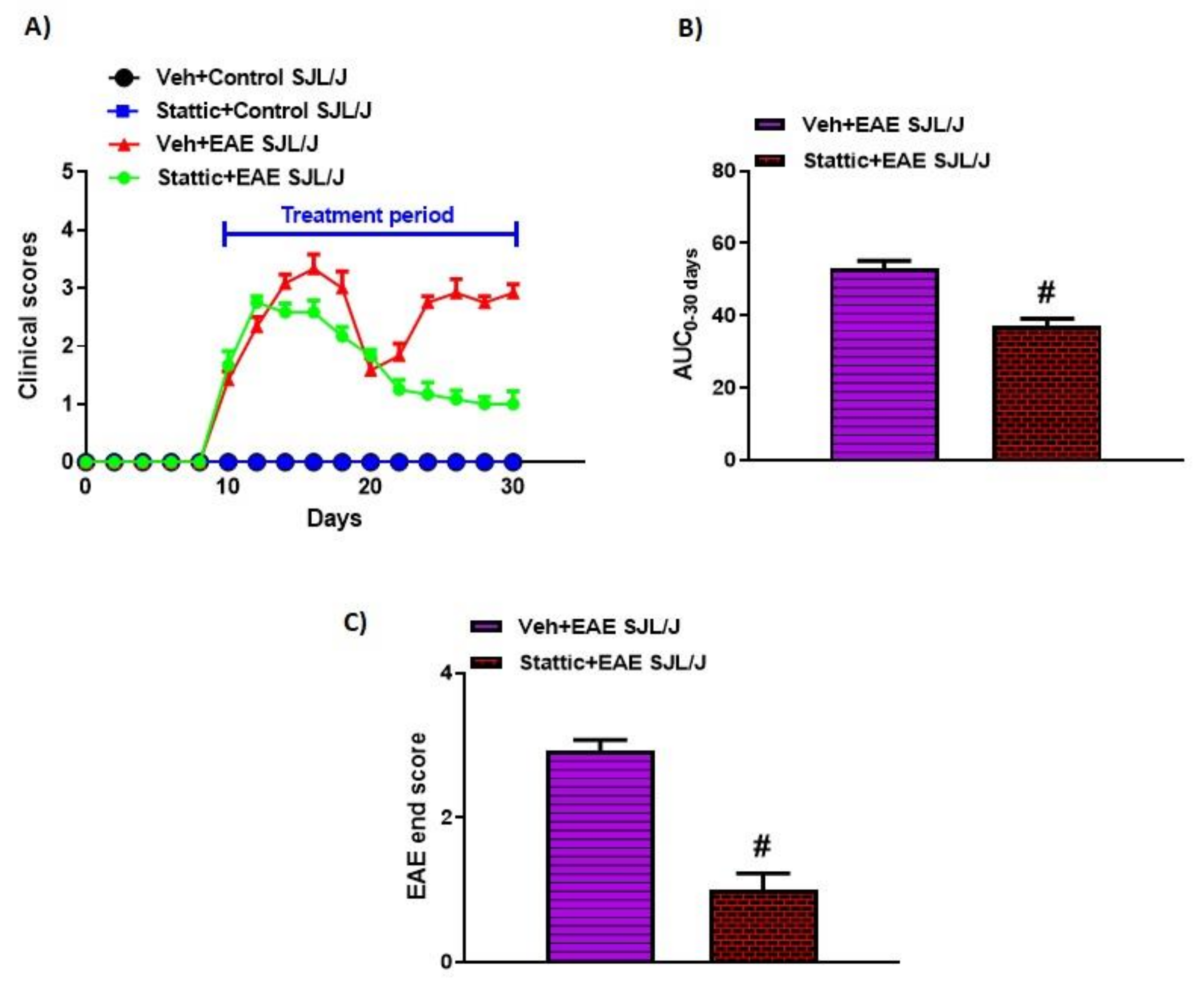{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Development of Relapsing–Remitting (RR) Experimental Autoimmune Encephalomyelitis (EAE) Model in SJL/J Mice
2.3. Experimental Groups
2.4. Real-Time PCR
2.5. Flow Cytometry of Cell Surface/Intracellular Markers in Splenocytes
2.6. Evaluation of Inflammatory Cytokines in the Brain by ELISA
2.7. Evaluation of Myeloperoxidase (MPO) Activity and Lipid Peroxides in the Brain
2.8. Evaluation of p-STAT3 Levels in the Brain by ELISA
2.9. Chemicals and Reagents
2.10. Statistical Analysis
3. Results
3.1. Blockade of STAT3 Signaling Improves Clinical Features Related to EAE in PLP-Immunized SJL/J Mice
3.2. Upregulation of Activated STAT3/Inflammatory Mediators and Its Attenuation by Stattic in Innate Immune Cells of PLP-Immunized SJL/J Mice
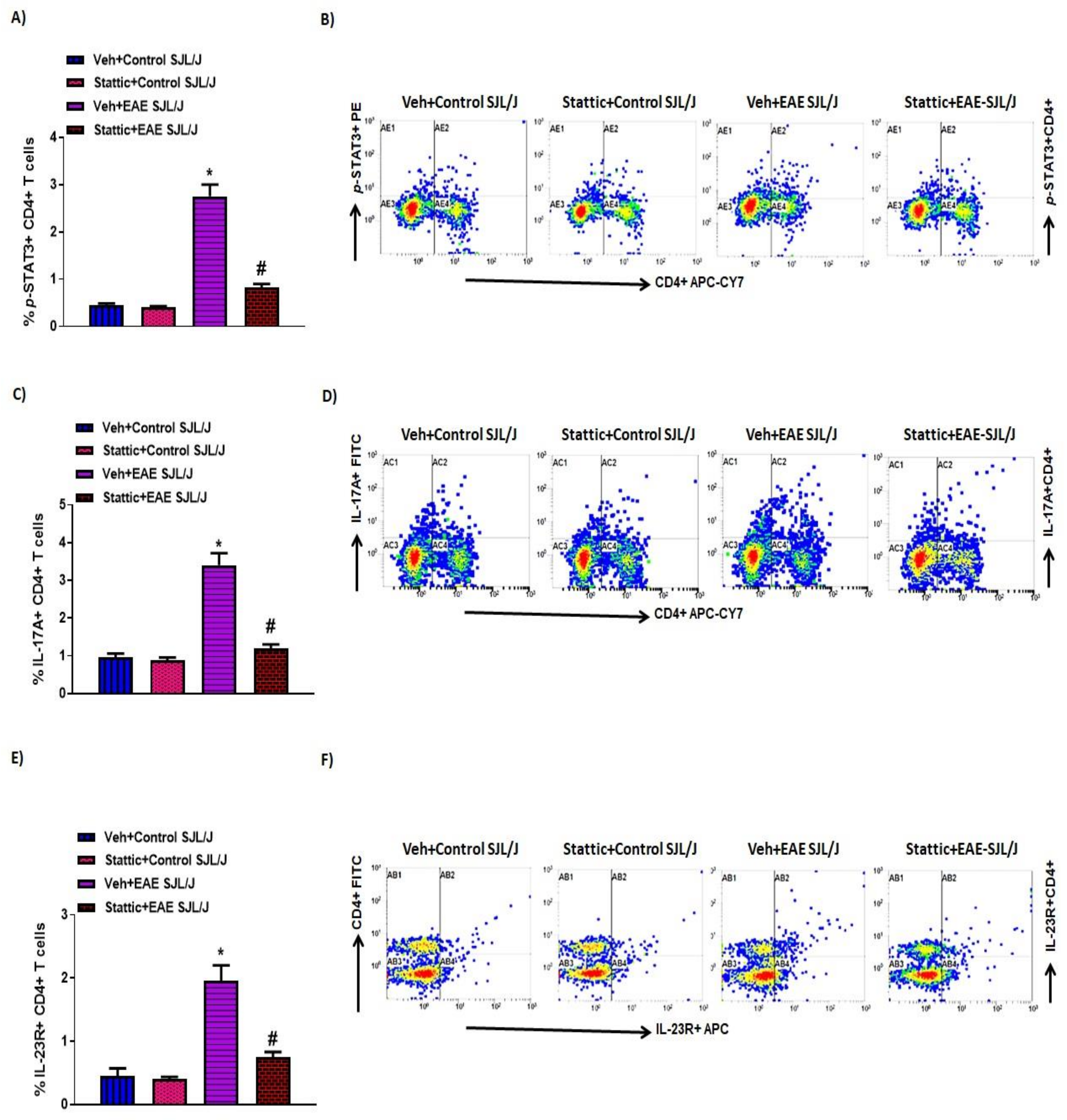
3.3. Upregulation of Activated STAT3/Inflammatory Mediators and Its Attenuation by Stattic in CD4+ T Cells in PLP-Immunized SJL/J Mice
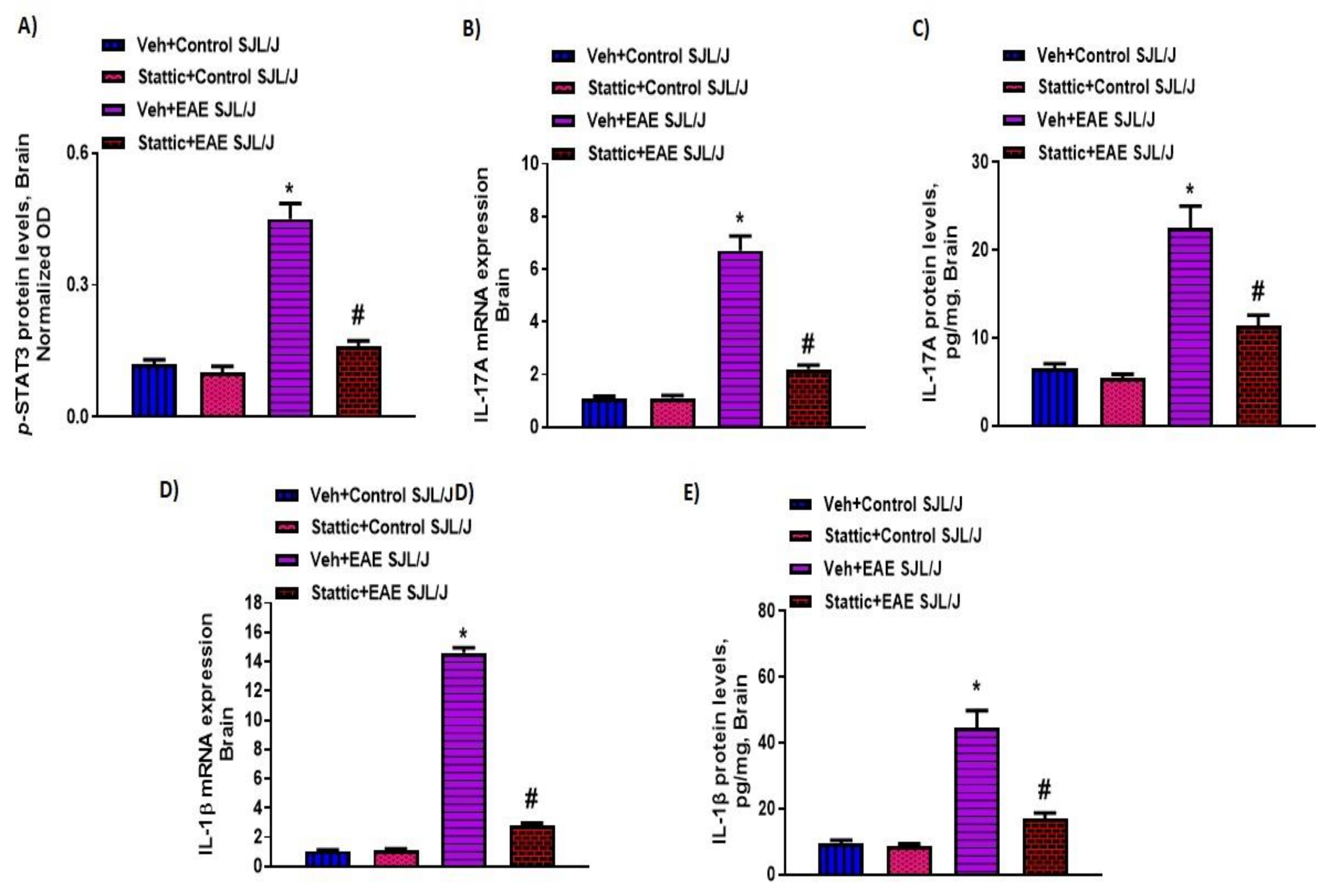
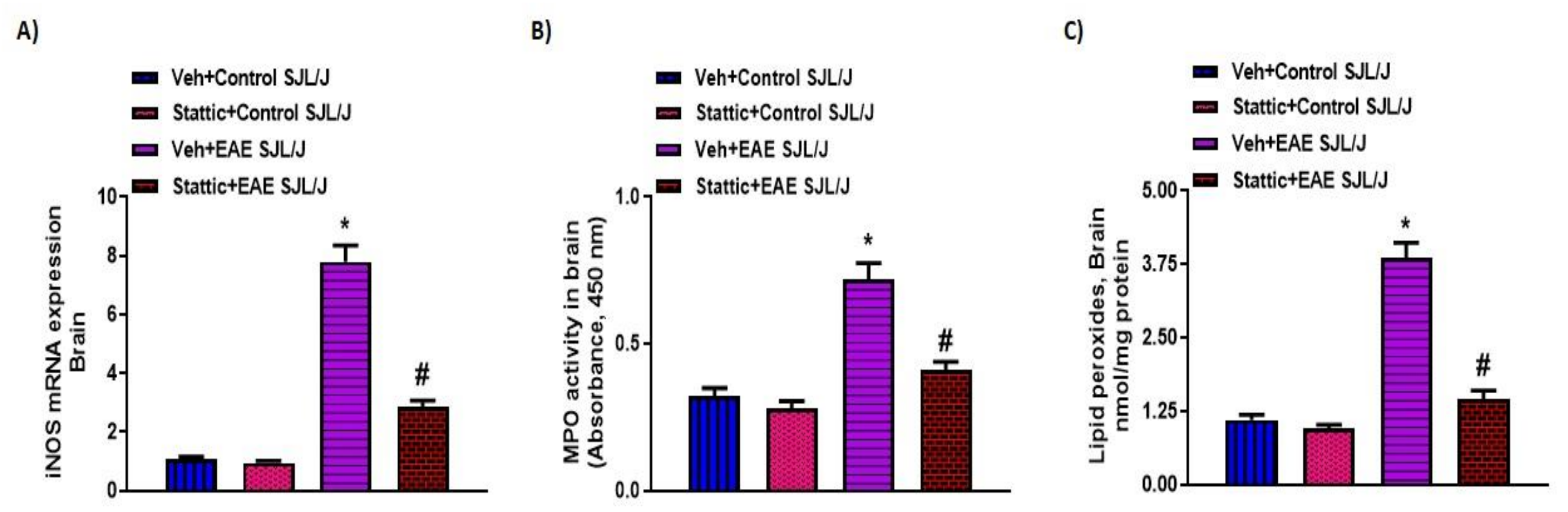
3.4. Upregulation of Activated STAT3, and Inflammatory/Oxidative Mediators, and Its Attenuation by Stattic in CNS of PLP-Immunized SJL/J Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achiron, A.; Gurevich, M.; Friedman, N.; Kaminski, N.; Mandel, M. Blood transcriptional signatures of multiple sclerosis: Unique gene expression of disease activity. Ann. Neurol. 2004, 55, 410–417. [Google Scholar] [CrossRef]
- Ratzer, R.; Søndergaard, H.B.; Christensen, J.R.; Börnsen, L.; Borup, R.; Sørensen, P.S.; Sellebjerg, F. Gene expression analysis of relapsing-remitting, primary progressive and secondary progressive multiple sclerosis. Mult. Scler. 2013, 19, 1841–1848. [Google Scholar] [CrossRef]
- Sonar, S.A.; Lal, G. Differentiation and transmigration of CD4 T cells in neuroinflammation and autoimmunity. Front. Immunol. 2017, 8, 1695. [Google Scholar] [CrossRef] [Green Version]
- Hertwig, L.; Pache, F.; Romero-Suarez, S.; Stürner, K.H.; Borisow, N.; Behrens, J.; Bellmann-Strobl, J.; Seeger, B.; Asselborn, N.; Ruprecht, K.; et al. Distinct functionality of neutrophils in multiple sclerosis and neuromyelitis optica. Mult. Scler. 2016, 22, 160–173. [Google Scholar] [CrossRef]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Locatelli, G.; Tai, Y.H.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; Groß, O.; Mai, D. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, 7554. [Google Scholar] [CrossRef]
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71. [Google Scholar] [CrossRef]
- Lu, H.C.; Kim, S.; Steelman, A.J.; Tracy, K.; Zhou, B.; Michaud, D.; Hillhouse, A.E.; Konganti, K.; Li, J. STAT3 signaling in myeloid cells promotes pathogenic myelin-specific T cell differentiation and autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2020, 117, 5430–5441. [Google Scholar] [CrossRef] [Green Version]
- Frisullo, G.; Nociti, V.; Iorio, R.; Patanella, A.K.; Marti, A.; Mirabella, M.; Tonali, P.A.; Batocchi, A.P. The persistency of high levels of pSTAT3 expression in circulating CD4+ T cells from CIS patients favors the early conversion to clinically defined multiple sclerosis. J. Neuroimmunol. 2008, 205, 126–134. [Google Scholar] [CrossRef]
- McGinley, A.M.; Sutton, C.E.; Edwards, S.C.; Leane, C.M.; de Courcey, J.; Teijeiro, A.; Hamilton, J.A.; Boon, L.; Djouder, N.; Mills, K.H. Interleukin-17A serves a priming, role in autoimmunity by recruiting, IL-1β-producing, myeloid cells that promote, pathogenic T Cells. Immunity 2020, 52, 342–356. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [Green Version]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Segal, B.M. Th17 cells in autoimmune demyelinating disease. Semin. Immunopathol. 2010, 32, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Lee, Y.S.; Yu, C.R.; Egwuagu, C.E. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J. Immunol. 2008, 180, 6070–6076. [Google Scholar] [CrossRef] [Green Version]
- Edwards, L.J.; Mizui, M.; Kyttaris, V. Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice. Clin. Immunol. 2015, 158, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Gavino, A.C.; Nahmod, K.; Bharadwaj, U.; Makedonas, G.; Tweardy, D.J. STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 2016, 71, 1684–1692. [Google Scholar] [CrossRef]
- Gharibi, T.; Babaloo, Z.; Hosseini, A.; Abdollahpour-Alitappeh, M.; Hashemi, V.; Marofi, F.; Nejati, K.; Baradaran, B. Targeting, STAT3 in cancer and autoimmune diseases. Eur. J. Pharmacol. 2020, 878, 173107. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018, 21, 541–551. [Google Scholar] [CrossRef]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Omachi, K.; Suico, M.A.; Kamura, M.; Kojima, H.; Fukuda, R.; Motomura, K.; Teramoto, K.; Kaseda, S.; Kuwazuru, J.; et al. STAT3 inhibition attenuates the progressive phenotypes of Alport syndrome mouse model. Nephrol. Dial. Transplant. 2018, 33, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; El-Sherbeeny, A.M.; Alasmari, A.F.; Alanazi, W.A.; Alasmari, F.; Ibrahim, K.E.; Al-Harbi, M.M.; Bakheet, S.A.; et al. Bruton’s tyrosine kinase inhibitor suppresses imiquimod-induced psoriasis-like inflammation in mice through regulation of IL-23/IL-17A in innate immune cells. Int. Immunopharmacol. 2020, 80, 106215. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Al-Harbi, N.O.; Ahmad, S.F.; Alhazzani, K.; Attia, S.M.; Alsanea, S.; Alhoshani, A.; Mahmood, H.M.; Alfardan, A.S.; Bakheet, S.A. Exposure to the plasticizer, Di-(2-ethylhexyl) phthalate during juvenile period exacerbates autism-like behavior in adult BTBR T + tf/J mice due to DNA hypomethylation and enhanced inflammation in brain and systemic immune cells. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 109, 110249. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta, Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; Bakheet, S.A.; El-Sherbeeny, A.M.; Ibrahim, K.E.; Alzahrani, K.S.; Al-Harbi, M.M.; Mahmood, H.M.; Alqahtani, F.; et al. Therapeutic treatment with Ibrutinib attenuates imiquimod-induced psoriasis-like inflammation in mice through downregulation of oxidative and inflammatory mediators in neutrophils and dendritic cells. Eur. J. Pharmacol. 2020, 877, 173088. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; El-Sherbeeny, A.M.; Al-Harbi, M.M.; Almukhlafi, T.S. GPR43 activation enhances psoriasis-like inflammation through epidermal upregulation of IL-6 and dual oxidase 2 signaling in a murine model. Cell. Signal 2017, 33, 59–68. [Google Scholar] [CrossRef]
- Bhat, R.; Steinman, L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron 2009, 64, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Paterka, M.; Siffrin, V.; Voss, J.O.; Werr, J.; Hoppmann, N.; Gollan, R.; Belikan, P.; Bruttger, J.; Birkenstock, J.; Jung, S.; et al. Gatekeeper role of brain antigen-presenting CD11c+ cells in neuroinflammation. EMBO J. 2016, 35, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.H.; Wang, Y.F.; He, D.D.; Zhang, X.M.; Zhou, Y.L.; Yue, H.; Huang, S.; Fu, Z.; Zhang, L.Y.; Mao, Z.Q.; et al. Let-7f-5p suppresses Th17 differentiation via targeting STAT3 in multiple sclerosis. Aging 2019, 11, 4463–4477. [Google Scholar] [CrossRef]
- Jakkula, E.; Leppä, V.; Sulonen, A.M.; Varilo, T.; Kallio, S.; Kemppinen, A.; Purcell, S.; Koivisto, K.; Tienari, P.; Sumelahti, M.L.; et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet. 2010, 86, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Lill, C.M.; Schjeide, B.M.; Akkad, D.A.; Blaschke, P.; Winkelmann, A.; Gerdes, L.A.; Hoffjan, S.; Luessi, F.; Dörner, T.; Li, S.C.; et al. Independent replication of STAT3 association with multiple sclerosis risk in a large German case-control sample. Neurogenetics 2012, 13, 83–86. [Google Scholar] [CrossRef]
- Frisullo, G.; Angelucci, F.; Caggiula, M.; Nociti, V.; Iorio, R.; Patanella, A.K.; Sancricca, C.; Mirabella, M.; Tonali, P.A.; Batocchi, A.P. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J. Neurosci. Res. 2006, 84, 1027–1036. [Google Scholar] [CrossRef]
- Canto, E.; Isobe, N.; Didonna, A.; Hauser, S.L.; Oksenberg, J.R.; MS-EPIC Study Group. Aberrant STAT phosphorylation signaling in peripheral blood mononuclear cells from multiple sclerosis patients. J. Neuroinflamm. 2018, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef]
- Baker, B.J.; Akhtar, L.N.; Benveniste, E.N. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 2009, 30, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Yeh, W.I.; De Sarno, P.; Holdbrooks, A.T.; Liu, Y.; Muldowney, M.T.; Reynolds, S.L.; Yanagisawa, L.L.; Fox, T.H.; Park, K.; et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 5004–5009. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Yang, W.; Parkitny, L.; Gibson, S.A.; Lee, K.S.; Collins, F.; Deshane, J.S.; Cheng, W.; Weinmann, A.S.; Wei, H.; et al. Deficiency of Socs3 leads to brain-targeted EAE via enhanced neutrophil activation and ROS production. JCI Insight 2019, 5, e126520. [Google Scholar] [CrossRef] [Green Version]
- Frisullo, G.; Mirabella, M.; Angelucci, F.; Caggiula, M.; Morosetti, R.; Sancricca, C.; Patanella, A.K.; Nociti, V.; Iorio, R.; Bianco, A.; et al. The effect of disease activity on leptin, leptin receptor and suppressor of cytokine signalling-3 expression in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2007, 192, 174–183. [Google Scholar] [CrossRef]
- Papenfuss, T.L.; Rogers, C.J.; Gienapp, I.; Yurrita, M.; McClain, M.; Damico, N.; Valo, J.; Song, F.; Whitacre, C.C. Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. J. Neuroimmunol. 2004, 150, 59–69. [Google Scholar] [CrossRef]
- Kumar, K.P.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.J.; Xu, Z.; Parthasarathy, U.; Drashansky, T.T.; Helm, E.Y.; Zuniga, A.N.; Lorentsen, K.J.; Mansouri, S.; Cho, J.Y.; Edelmann, M.J.; et al. Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation. Nat. Commun. 2019, 10, 701. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Lévesque, S.A.; Paré, A.; Mailhot, B.; Bellver-Landete, V.; Kébir, H.; Lécuyer, M.A.; Alvarez, J.I.; Prat, A.; Vaccari, J.P.D.R.; Keane, R.W.; et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med. 2016, 213, 929–949. [Google Scholar] [CrossRef]
- Zakrzewska-Pniewska, B.; Styczynska, M.; Podlecka, A.; Samocka, R.; Peplonska, B.; Barcikowska, M.; Kwiecinski, H. Association of apolipoprotein E and myeloperoxidase genotypes to clinical course of familial and sporadic multiple sclerosis. Mult. Scler. 2004, 10, 266–271. [Google Scholar] [CrossRef]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2016, 136, 826–836. [Google Scholar] [CrossRef] [Green Version]
- De Bondt, M.; Hellings, N.; Opdenakker, G.; Struyf, S. Neutrophils: Underestimated, players in the pathogenesis of multiple, sclerosis (MS). Int. J. Mol. Sci. 2020, 21, 4558. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Bevilacqua, D.; Cassatella, M.A.; Scapini, P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019, 156, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbach, K.; Piedavent, M.; Bauer, S.; Neumann, J.T.; Friese, M.A. Neutrophils amplify autoimmune central nervous system infiltrates by maturing local APCs. J. Immunol 2013, 191, 4531–4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeem, A.; Siddiqui, N.; Alharbi, N.O.; Alharbi, M.M. Airway and systemic oxidant-antioxidant dysregulation in asthma: A possible scenario of oxidants spill over from lung into blood. Pulm. Pharmacol. Ther. 2014, 29, 31–40. [Google Scholar] [CrossRef]
- Li, W.; Wu, H.; Gao, C.; Yang, D.; Yang, D.; Shen, J. Radix, Rehmanniae Extract, Ameliorates Experimental, Autoimmune Encephalomyelitis by Suppressing, Macrophage-Derived, Nitrative Damage. Front. Physiol. 2018, 9, 864. [Google Scholar] [CrossRef]
- Cong, H.; Zhang, M.; Chang, H.; Du, L.; Zhang, X.; Yin, L. Icariin ameliorates the progression of experimental autoimmune encephalomyelitis by down-regulating the major inflammatory signal pathways in a mouse relapse-remission model of multiple sclerosis. Eur. J. Pharmacol. 2020, 885, 173523. [Google Scholar] [CrossRef]
- Lin, C.C.; Edelson, B.T. New, Insights into the Role of IL-1β in Experimental, Autoimmune Encephalomyelitis and Multiple, Sclerosis. J. Immunol. 2017, 198, 4553–4560. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhazzani, K.; Ahmad, S.F.; Al-Harbi, N.O.; Attia, S.M.; Bakheet, S.A.; Sarawi, W.; Alqarni, S.A.; Algahtani, M.; Nadeem, A. Pharmacological Inhibition of STAT3 by Stattic Ameliorates Clinical Symptoms and Reduces Autoinflammation in Myeloid, Lymphoid, and Neuronal Tissue Compartments in Relapsing–Remitting Model of Experimental Autoimmune Encephalomyelitis in SJL/J Mice. Pharmaceutics 2021, 13, 925. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13070925
Alhazzani K, Ahmad SF, Al-Harbi NO, Attia SM, Bakheet SA, Sarawi W, Alqarni SA, Algahtani M, Nadeem A. Pharmacological Inhibition of STAT3 by Stattic Ameliorates Clinical Symptoms and Reduces Autoinflammation in Myeloid, Lymphoid, and Neuronal Tissue Compartments in Relapsing–Remitting Model of Experimental Autoimmune Encephalomyelitis in SJL/J Mice. Pharmaceutics. 2021; 13(7):925. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13070925
Chicago/Turabian StyleAlhazzani, Khalid, Sheikh F. Ahmad, Naif O. Al-Harbi, Sabry M. Attia, Saleh A. Bakheet, Wedad Sarawi, Saleh A. Alqarni, Mohammad Algahtani, and Ahmed Nadeem. 2021. "Pharmacological Inhibition of STAT3 by Stattic Ameliorates Clinical Symptoms and Reduces Autoinflammation in Myeloid, Lymphoid, and Neuronal Tissue Compartments in Relapsing–Remitting Model of Experimental Autoimmune Encephalomyelitis in SJL/J Mice" Pharmaceutics 13, no. 7: 925. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13070925