
Optimization and Development of Selective Histone Deacetylase Inhibitor (MPT0B291)-Loaded Albumin Nanoparticles for Anticancer Therapy

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of MPT0B291-Loaded Albumin NPs
2.2. Particle Size, Size Distribution, Zeta Potential, and Surface Morphology
2.3. Encapsulation Efficiency (EE) and Drug Loading (DL) of MPT0B291-HSA NPs
2.4. Stability of MPT0B291-HSA NPs
2.5. In Vitro Drug-Release Behavior
2.6. Cell Viability Assay
2.7. Maximum Tolerated Dose (MTD)
2.8. In Vivo PK Study
2.9. Statistical Analysis
3. Results and Discussion
3.1. Optimization and Preparation of HSA NPs
3.1.1. Effect of the Energy Output of Ultrasonication
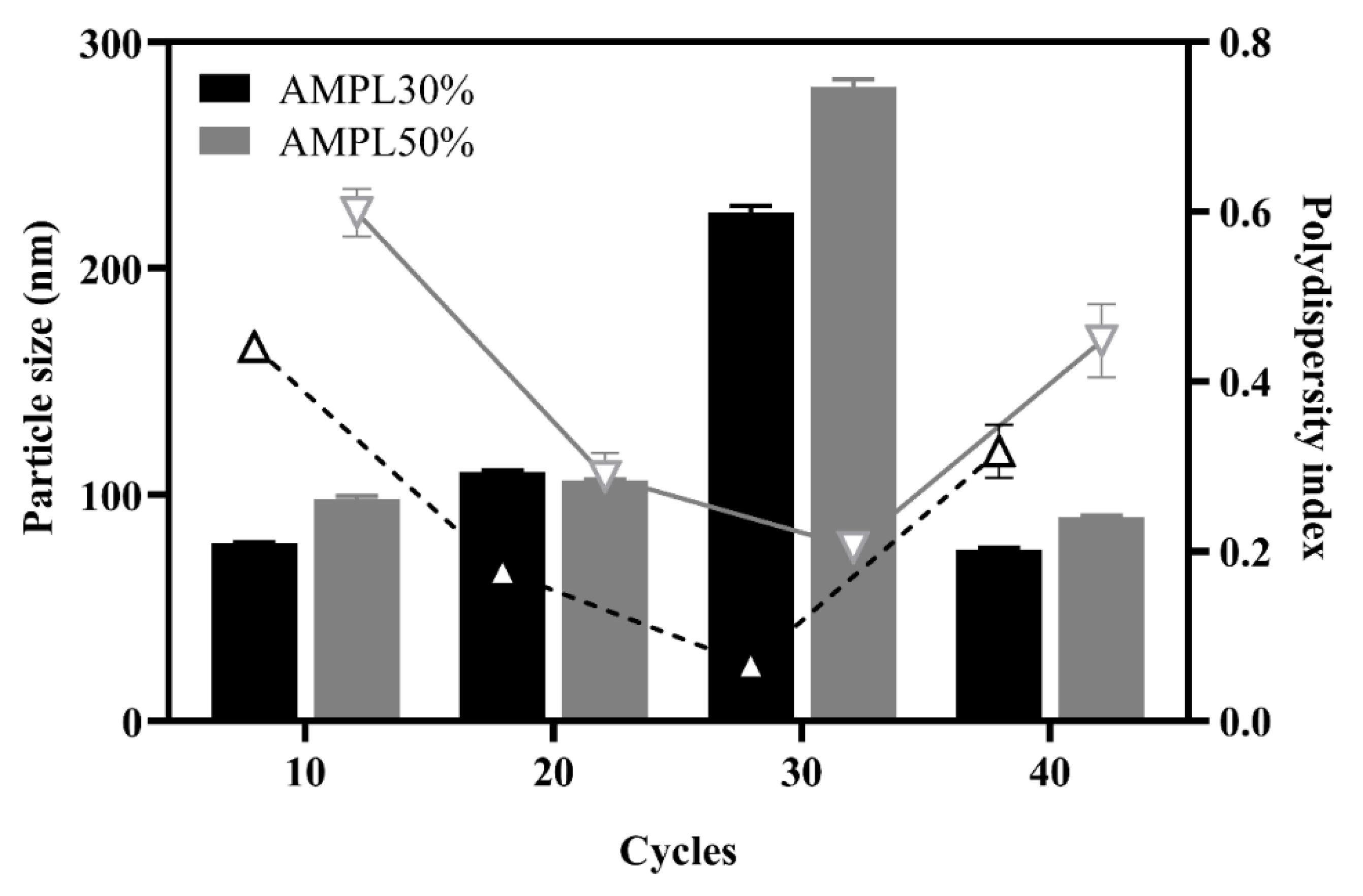
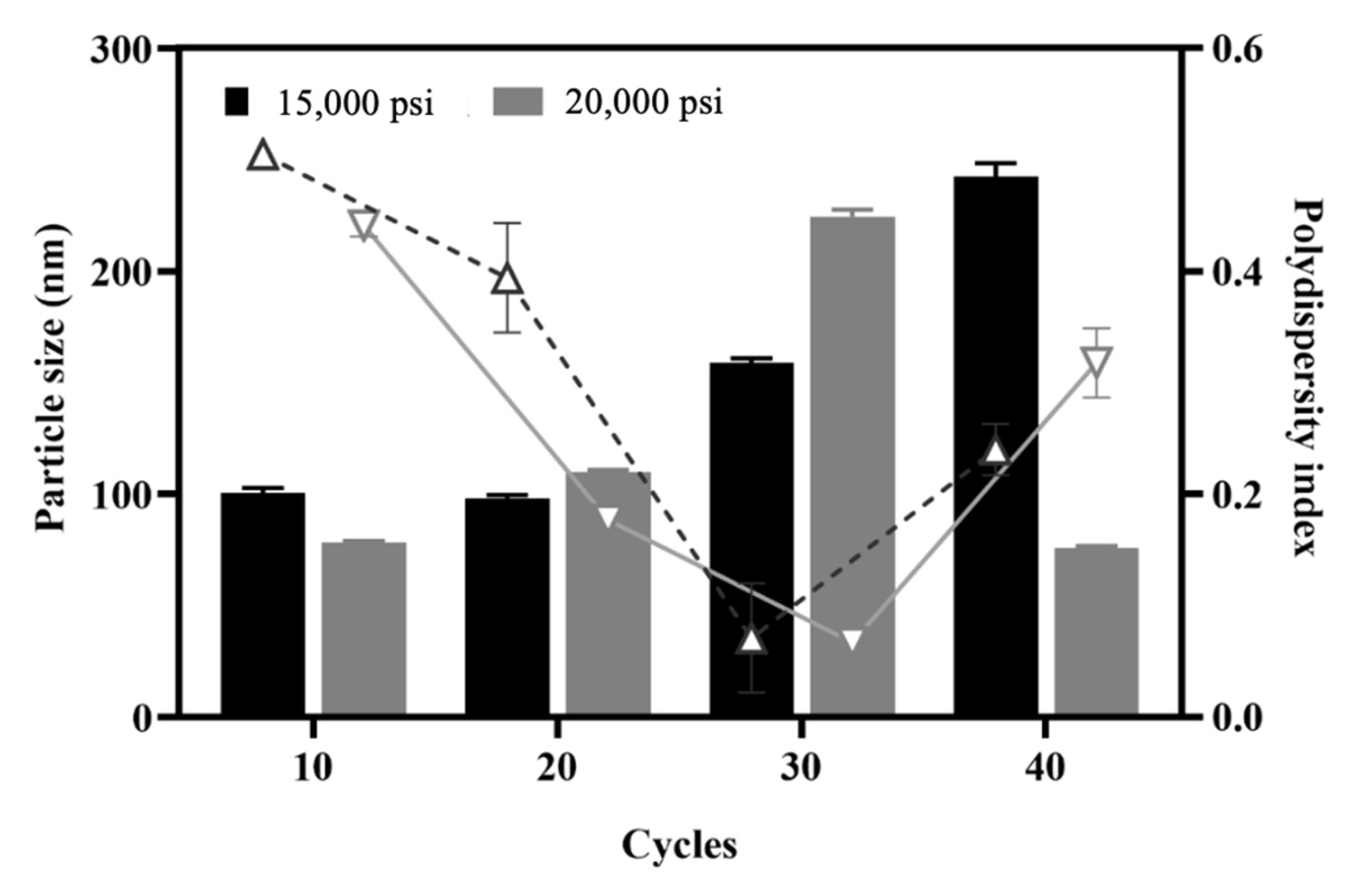
3.1.2. Effect of the Number of Homogenization Cycles and Pressure
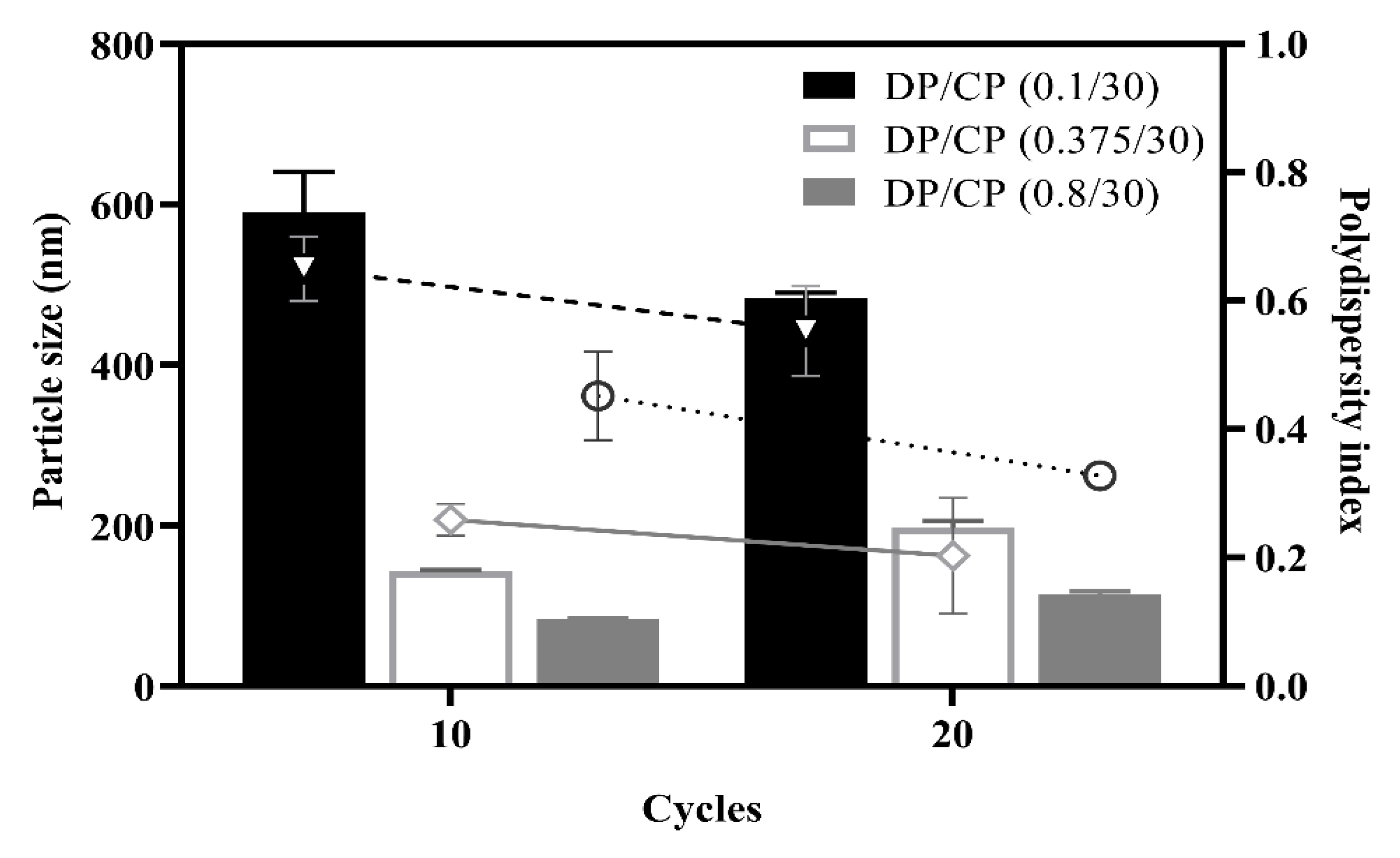
3.1.3. Effect of the Ratio of Dispersed Phase (DP) to Continuous Phase (CP)
3.2. Characterization of Albumin NPs and MPT0B291-HSA NPs
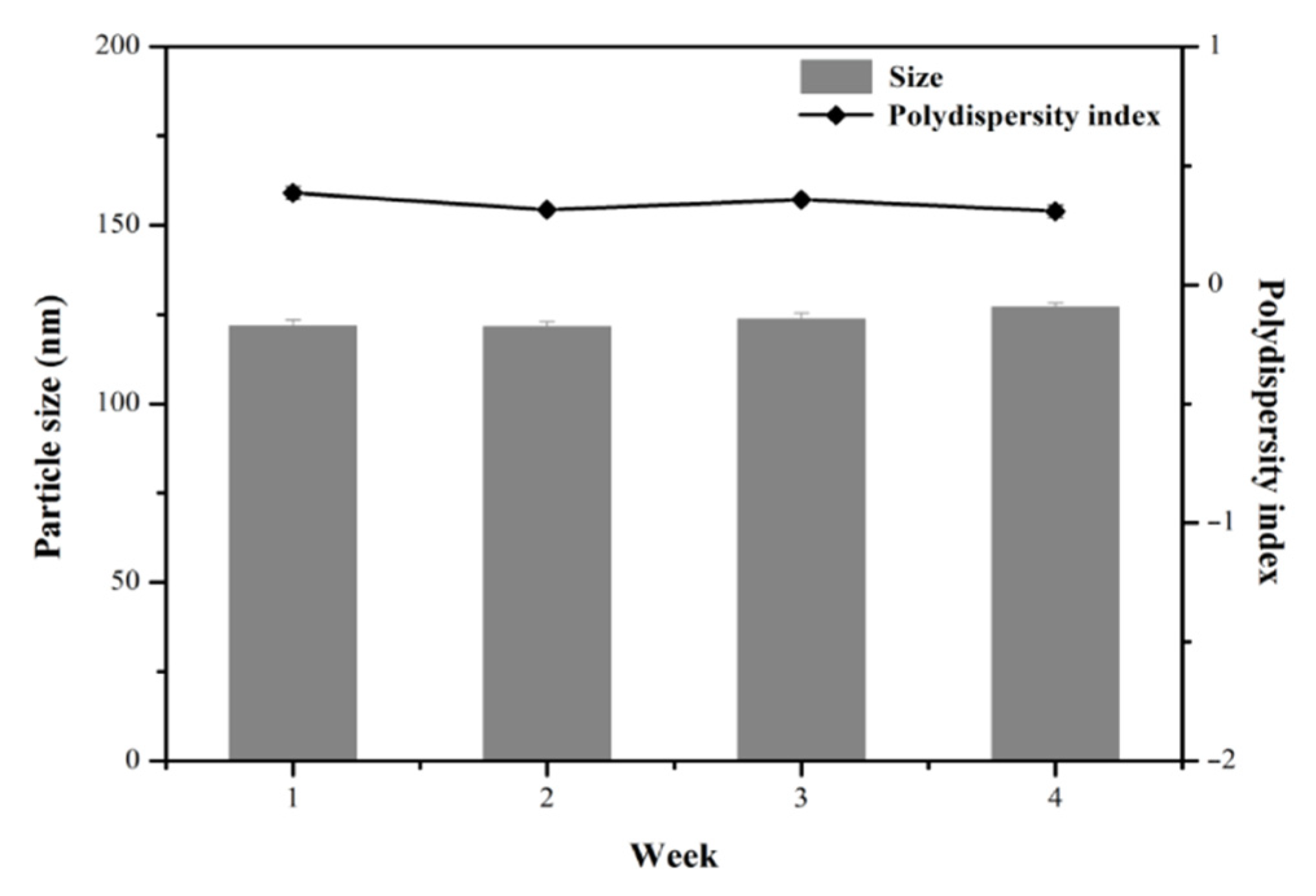
3.3. Stability Studies of MPT0B291-HSA NPs
3.4. In Vitro Drug Release
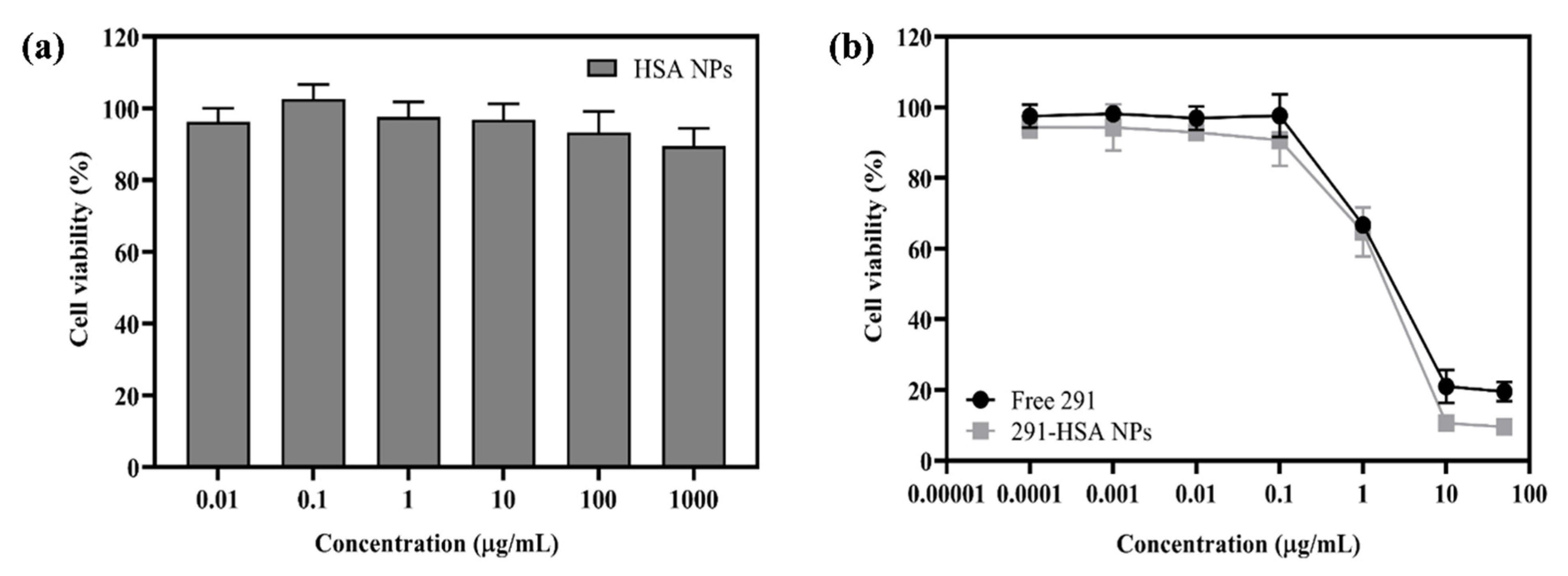
3.5. In Vitro Cytotoxicity Test
3.6. Maximum Tolerated Dose (MTD)
3.7. In Vivo PK Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Peserico, A.; Simone, C. Physical and functional hat/hdac interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of acetylation at lys16 and trimethylation at lys20 of histone h4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Yasui, W.; Oue, N.; Ono, S.; Mitani, Y.; Ito, R.; Nakayama, H. Histone acetylation and gastrointestinal carcinogenesis. Ann. N. Y. Acad. Sci. 2003, 983, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (hdacs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of hdac6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A. Discovery and development of saha as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Grabarska, A.; Łuszczki, J.J.; Nowosadzka, E.; Gumbarewicz, E.; Jeleniewicz, W.; Dmoszyńska-Graniczka, M.; Kowalczuk, K.; Kupisz, K.; Polberg, K.; Stepulak, A. Histone deacetylase inhibitor saha as potential targeted therapy agent for larynx cancer cells. J. Cancer 2017, 8, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual zinc-binding mode of hdac6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Yue, Y.; Pan, M.; Sun, J.; Chu, J.; Lin, X.; Xu, W.; Feng, L.; Chen, Y.; Chen, D. Histone deacetylase 3 inhibits new tumor suppressor gene dtwd1 in gastric cancer. Am. J. Cancer Res. 2015, 5, 663. [Google Scholar]
- Deng, B.; Luo, Q.; Halim, A.; Liu, Q.; Zhang, B.; Song, G. The Antiangiogenesis Role of Histone Deacetylase Inhibitors: Their Potential Application to Tumor Therapy and Tissue Repair. DNA Cell Biol. 2020, 39, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Buyandelger, B.; Bar, E.E.; Hung, K.S.; Chen, R.M.; Chiang, Y.H.; Liou, J.P.; Huang, H.M.; Wang, J.Y. Histone deacetylase inhibitor mpt0b291 suppresses glioma growth in vitro and in vivo partially through acetylation of p53. Int. J. Biol. Sci. 2020, 16, 3184–3199. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Tsai, A.C.; Chen, M.C.; Shen, P.J.; Cheng, Y.C.; Kuo, C.C.; Pan, S.L.; Liu, Y.M.; Liu, J.F.; Yeh, T.K.; et al. Azaindolylsulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer hct116 cells. J. Med. Chem. 2014, 57, 4009–4022. [Google Scholar] [CrossRef] [PubMed]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahann, J. Protein nanoparticles as multifunctional drug delivery carriers. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2019; Volume 257. [Google Scholar]
- Lohcharoenkal, W.; Wang, L.Y.; Chen, Y.C.; Rojanasakul, Y. Protein nanoparticles as drug delivery carriers for cancer therapy. Biomed. Res. Int. 2014, 2014, 180549. [Google Scholar] [CrossRef] [Green Version]
- Desai, N. Nanoparticle albumin-bound paclitaxel (abraxane®). In Albumin in Medicine; Springer: Singapore, 2016; pp. 101–119. [Google Scholar]
- Yin, T.J.; Dong, L.H.; Cui, B.; Wang, L.; Yin, L.F.; Zhou, J.P.; Huo, M.R. A toxic organic solvent-free technology for the preparation of pegylated paclitaxel nanosuspension based on human serum albumin for effective cancer therapy. Int. J. Nanomed. 2015, 10, 7397–7412. [Google Scholar]
- Tai, C.J.; Wang, H.; Wang, C.K.; Tai, C.J.; Huang, M.T.; Wu, C.H.; Chen, R.J.; Kuo, L.J.; Wei, P.L.; Chang, Y.J.; et al. Bevacizumab and cetuximab with conventional chemotherapy reduced pancreatic tumor weight in mouse pancreatic cancer xenografts. Clin. Exp. Med. 2017, 17, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Thadakapally, R.; Aafreen, A.; Aukunuru, J.; Habibuddin, M.; Jogala, S. Preparation and characterization of peg-albumin-curcumin nanoparticles intended to treat breast cancer. Indian J. Pharm. Sci. 2016, 78, 65–72. [Google Scholar]
- Shao, H.; Tang, H.; Salavaggione, O.E.; Yu, C.; Hylander, B.; Tan, W.; Repasky, E.; Adjei, A.A.; Dy, G.K. Improved response to nab-paclitaxel compared with cremophor-solubilized paclitaxel is independent of secreted protein acidic and rich in cysteine expression in non-small cell lung cancer. J. Thorac Oncol. 2011, 6, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; Tong, Z.; Hou, S.; Bateman, S.; Pierce, D.W.; Heise, C.; Von Hoff, D.D.; Maitra, A.; et al. Superior therapeutic efficacy of nab-paclitaxel over cremophor-based paclitaxel in locally advanced and metastatic models of human pancreatic cancer. Br. J. Cancer 2016, 115, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef]
- Peng, Q.; Mu, H. The potential of protein-nanomaterial interaction for advanced drug delivery. J. Control. Release 2016, 225, 121–132. [Google Scholar] [CrossRef]
- Karimi, M.; Bahrami, S.; Ravari, S.B.; Zangabad, P.S.; Mirshekari, H.; Bozorgomid, M.; Shahreza, S.; Sori, M.; Hamblin, M.R. Albumin nanostructures as advanced drug delivery systems. Expert Opin. Drug Deliv. 2016, 13, 1609–1623. [Google Scholar] [CrossRef] [Green Version]
- Food Drug Administration, U.S. Bioanalytical Method Validation Guidance for Industry; US Department of Health Human Services: Washington, DC, USA, 2018; pp. 1–41.
- Tang, B.; Fang, G.; Gao, Y.; Liu, Y.; Liu, J.; Zou, M.; Cheng, G. Liprosomes loading paclitaxel for brain-targeting delivery by intravenous administration: In vitro characterization and in vivo evaluation. Int. J. Pharm. 2014, 475, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Eisenberg, A. Polymer vesicles. Science 2002, 297, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichhane, S.; Lee, S. Albumin nanoscience: Homing nanotechnology enabling targeted drug delivery and therapy. Arch. Pharm. Res. 2020, 43, 118–133. [Google Scholar] [CrossRef]
- Jun, J.Y.; Nguyen, H.H.; Paik, S.-Y.-R.; Chun, H.S.; Kang, B.-C.; Ko, S. Preparation of size-controlled bovine serum albumin (bsa) nanoparticles by a modified desolvation method. Food Chem. 2011, 127, 1892–1898. [Google Scholar] [CrossRef]
- Lomis, N.; Westfall, S.; Farahdel, L.; Malhotra, M.; Shum-Tim, D.; Prakash, S. Human serum albumin nanoparticles for use in cancer drug delivery: Process optimization and in vitro characterization. Nanomaterials 2016, 6, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazhbayev, Y.; Mukashev, O.; Burkeev, M.; Kreuter, J. Hydroxyurea-loaded albumin nanoparticles: Preparation, characterization, and in vitro studies. Pharmaceutics 2019, 11, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Thareja, S. In vitro and in vivo characterization of pharmaceutical nanocarriers used for drug delivery. Artif. Cells Nanomed. Biotechnol. 2019, 47, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C.-X. Development of high-drug-loading nanoparticles. ChemPlusChem 2020, 85, 2143–2157. [Google Scholar] [CrossRef] [PubMed]
- Gronroos, A.; Pirkonen, P.; Heikkinen, J.; Ihalainen, J.; Mursunen, H.; Sekki, H. Ultrasonic depolymerization of aqueous polyvinyl alcohol. Ultrason. Sonochem. 2001, 8, 259–264. [Google Scholar] [CrossRef]
- Amini, M.A.; Faramarzi, M.A.; Mohammadyani, D.; Esmaeilzadeh-Gharehdaghi, E.; Amani, A. Modeling the parameters involved in preparation of pla nanoparticles carrying hydrophobic drug molecules using artificial neural networks. J. Pharm. Innov. 2013, 8, 111–120. [Google Scholar] [CrossRef]
- Stinchcombe, T.E. Nanoparticle albumin-bound paclitaxel: A novel cremphor-el-free formulation of paclitaxel. Nanomedicine 2007, 2, 415–423. [Google Scholar] [CrossRef]
- Mohan, S.; Narsimhan, G. Coalescence of protein-stabilized emulsions in a high-pressure homogenizer. J. Colloid Interface Sci. 1997, 192, 1–15. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, X.; Zu, Y.; Zhang, Y. Preparation and characterization of paclitaxel nanosuspension using novel emulsification method by combining high speed homogenizer and high pressure homogenization. Int. J. Pharm. 2015, 490, 324–333. [Google Scholar] [CrossRef]
- Mu, L.; Feng, S.S. A novel controlled release formulation for the anticancer drug paclitaxel (taxol): Plga nanoparticles containing vitamin E TPGS. J. Control. Release 2003, 86, 33–48. [Google Scholar] [CrossRef]
- Jaramillo, D.P.; Roberts, R.F.; Coupland, J.N. Effect of ph on the properties of soy protein–pectin complexes. Food Res. Int. 2011, 44, 911–916. [Google Scholar] [CrossRef]
- De Oliveira, J.K.; Ronik, D.F.; Ascari, J.; Mainardes, R.M.; Khalil, N.M. Nanoencapsulation of apocynin in bovine serum albumin nanoparticles: Physicochemical characterization. Nanosci. Nanotechnol. Asia 2018, 8, 90–99. [Google Scholar] [CrossRef]
- He, X.; Xiang, N.; Zhang, J.; Zhou, J.; Fu, Y.; Gong, T.; Zhang, Z. Encapsulation of teniposide into albumin nanoparticles with greatly lowered toxicity and enhanced antitumor activity. Int. J. Pharm. 2015, 487, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Sun, Y.; Li, Y.; Hao, F.; Qiu, P.; Teng, L.; Xie, J.; Gao, Y. Docetaxel-loaded human serum albumin (HSA) nanoparticles: Synthesis, characterization, and evaluation. Biomed. Eng. Online 2019, 18, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Ma, K.; Kim, T.H.; Lee, E.S.; Oh, K.T.; Park, E.S.; Lee, K.C.; Youn, Y.S. Doxorubicin-loaded human serum albumin nanoparticles surface-modified with tnf-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types. Biomaterials 2012, 33, 1536–1546. [Google Scholar] [CrossRef]
- Ogawara, K.-I.; Furumoto, K.; Nagayama, S.; Minato, K.; Higaki, K.; Kai, T.; Kimura, T. Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: Implications for rational design of nanoparticles. J. Control. Release 2004, 100, 451–455. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Size (nm) | PDI | Zeta Potential (mV) | EE (%) | DL (%) | Albumin Recovery (%) | Ratio of Albumin to Drug | |
|---|---|---|---|---|---|---|---|
| MPT0B291-HSA NPs | 136.3 ± 4.51 | 0.241 ± 0.077 | −1.87 ± 0.133 | 92.0 ± 1.11 | 5.97 ± 0.07 | 95.0 ± 1.24 | 9.6 |
| Parameter | MPT0B291-HSA NPs | MPT0B291 Solution |
|---|---|---|
| AUC0–24h (h⋅ng/mL) | 2225.09 ± 767.70 ** | 290.98 ± 39.73 |
| AUC0–∞ (h⋅ng/mL) | 2333.73 ± 764.32 ** | 292.60 ± 39.17 |
| t1/2 (h) | 4.71 ± 0.65 ** | 2.07 ± 0.64 |
| CL (mL/min/kg) | 38.24 ± 10.53 *** | 288.47 ± 36.66 |
| MRT (h) | 4.95 ± 1.03 ** | 1.86 ± 0.66 |
| Vss (L/kg) | 11.49 ± 3.92 | 32.58 ± 12.26 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putri, A.D.; Chen, P.-S.; Su, Y.-L.; Lin, J.-P.; Liou, J.-P.; Hsieh, C.-M. Optimization and Development of Selective Histone Deacetylase Inhibitor (MPT0B291)-Loaded Albumin Nanoparticles for Anticancer Therapy. Pharmaceutics 2021, 13, 1728. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13101728
Putri AD, Chen P-S, Su Y-L, Lin J-P, Liou J-P, Hsieh C-M. Optimization and Development of Selective Histone Deacetylase Inhibitor (MPT0B291)-Loaded Albumin Nanoparticles for Anticancer Therapy. Pharmaceutics. 2021; 13(10):1728. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13101728
Chicago/Turabian StylePutri, Athika Darumas, Pai-Shan Chen, Yu-Lin Su, Jia-Pei Lin, Jing-Ping Liou, and Chien-Ming Hsieh. 2021. "Optimization and Development of Selective Histone Deacetylase Inhibitor (MPT0B291)-Loaded Albumin Nanoparticles for Anticancer Therapy" Pharmaceutics 13, no. 10: 1728. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13101728