
Design, Synthesis, and Evaluation of Linker-Optimised PSMA-Targeting Radioligands

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
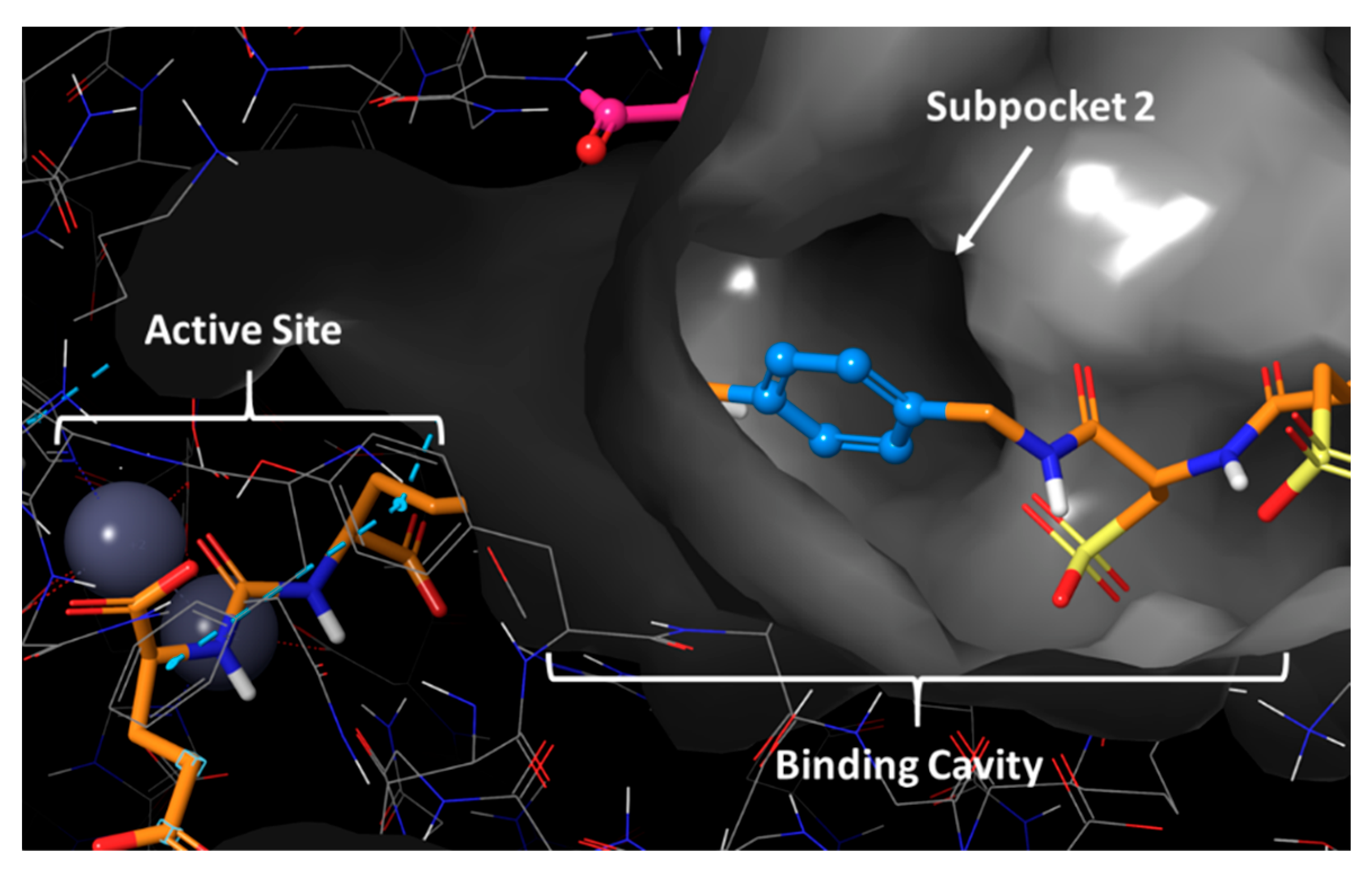
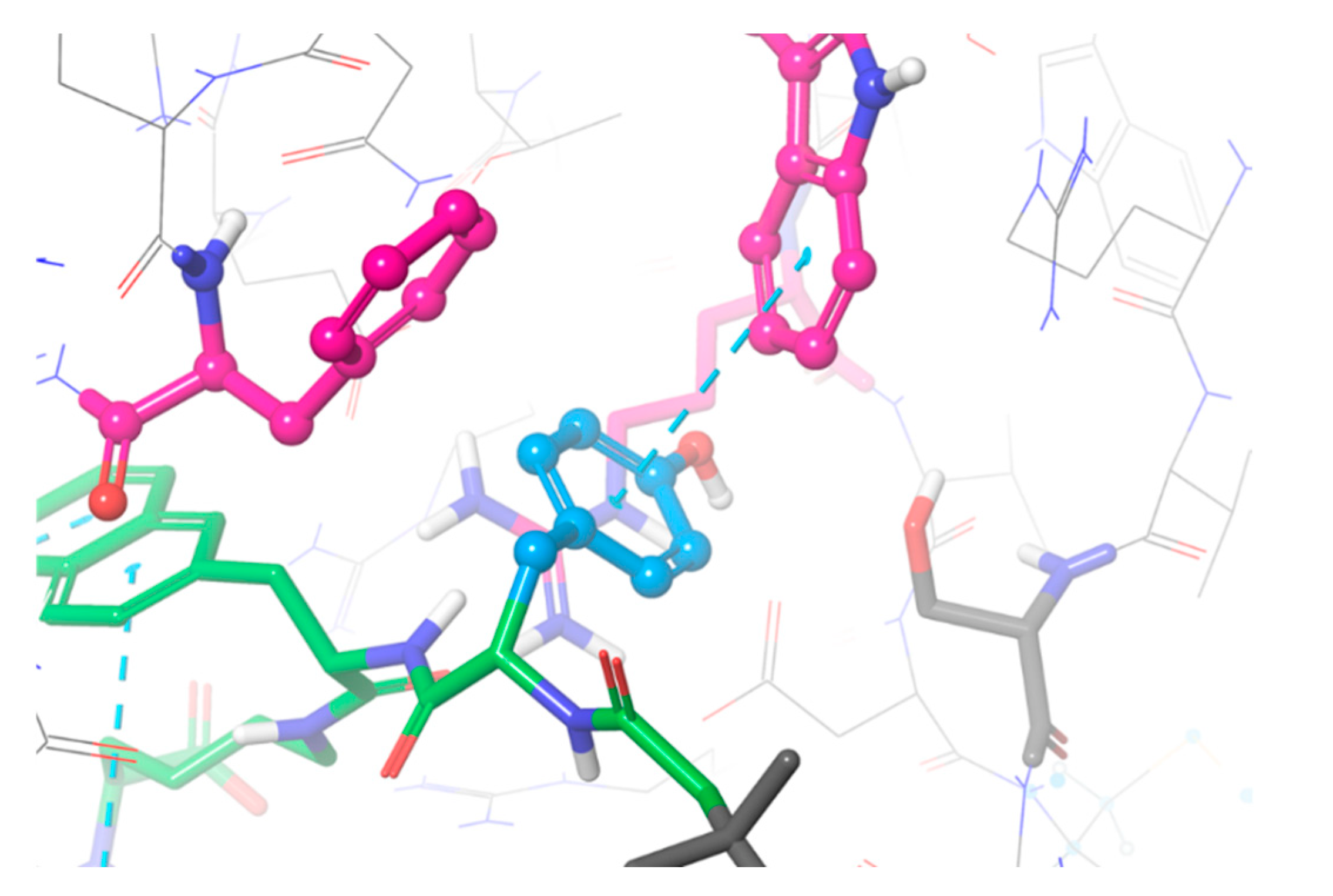
2.1. Computational Modelling
2.2. Synthesis
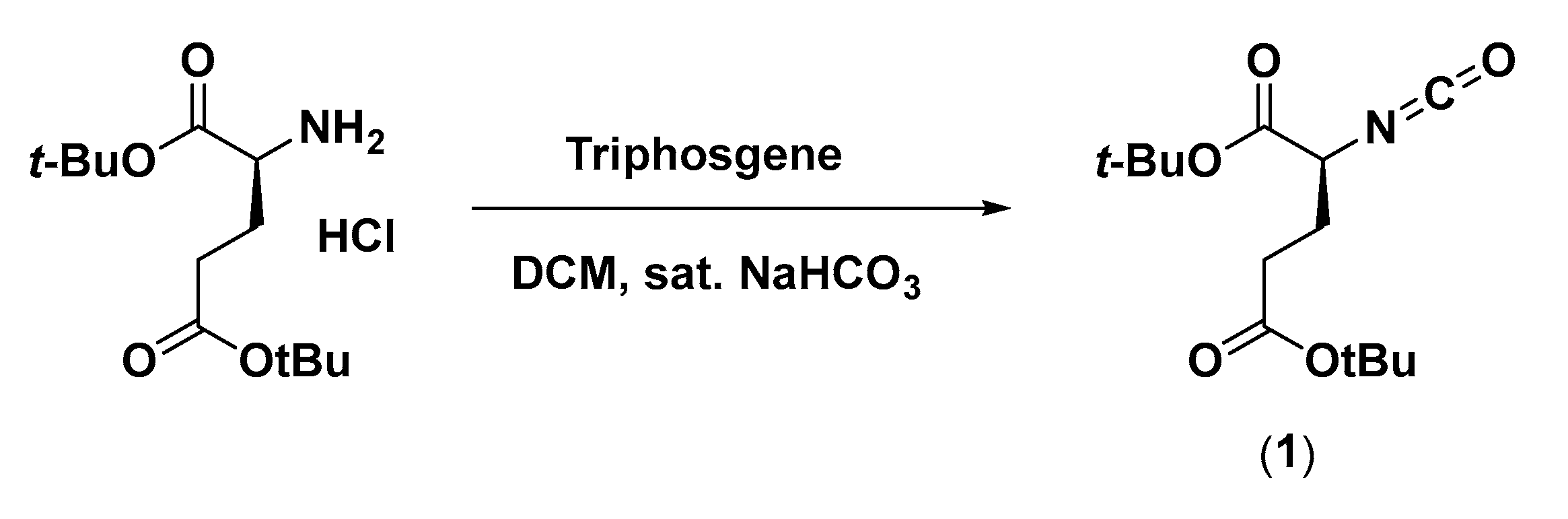
2.2.1. Di-tert-butyl 2-isocyanatopentanedioate (1)
2.2.2. Di-tert-butyl (((S)-6-amino-1-methoxy-1-oxohexan-2-yl)carbamoyl)-L-glutamate (2)

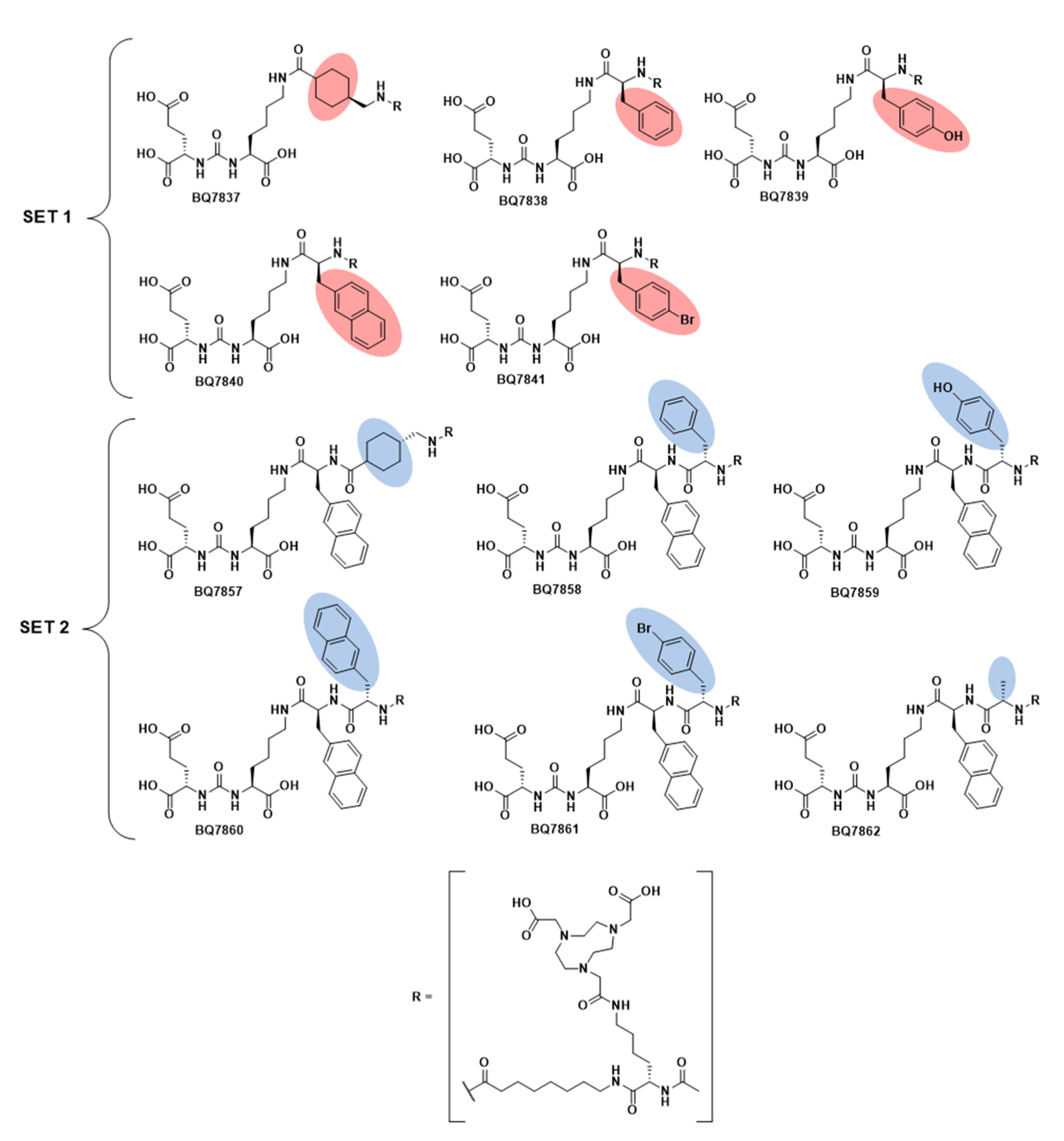
2.2.3. General Procedure for BQ7837-41 (SET 1)
BQ7837
BQ7838
BQ7839
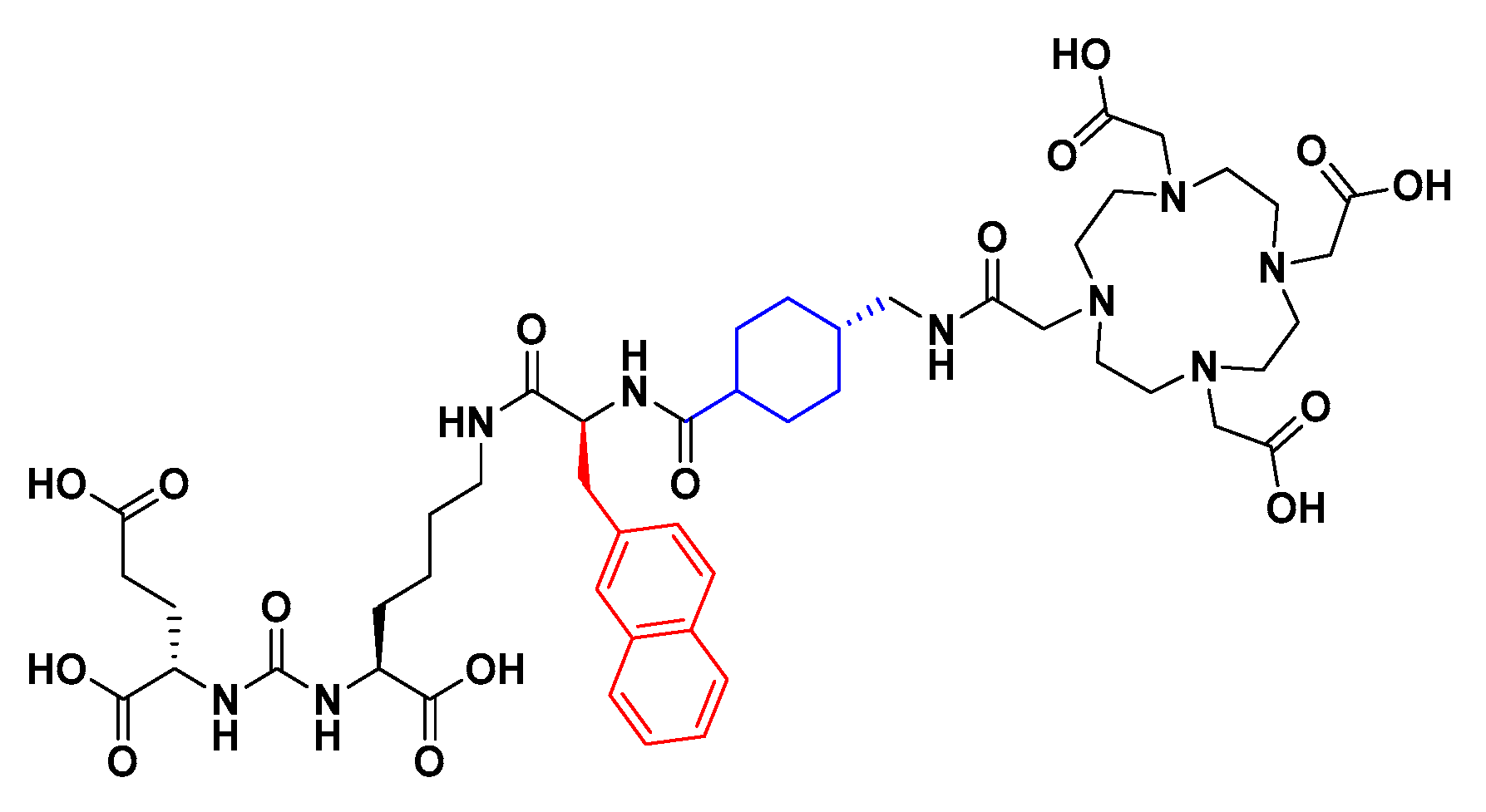
BQ7840
BQ7841
2.2.4. Di-tert-butyl (((S)-6-((S)-2-amino-3-(naphthalen-2-yl)propanamido)-1-methoxy-1-oxohexan-2-yl)carbamoyl)-L-glutamate (3)

2.2.5. General Procedure for BQ7857-62 (SET 2)
BQ7857
BQ7858
BQ7859
BQ7860
BQ7861
BQ7862
2.3. Radiochemistry
2.4. Octanol/Water
2.5. In Vitro Characterisation
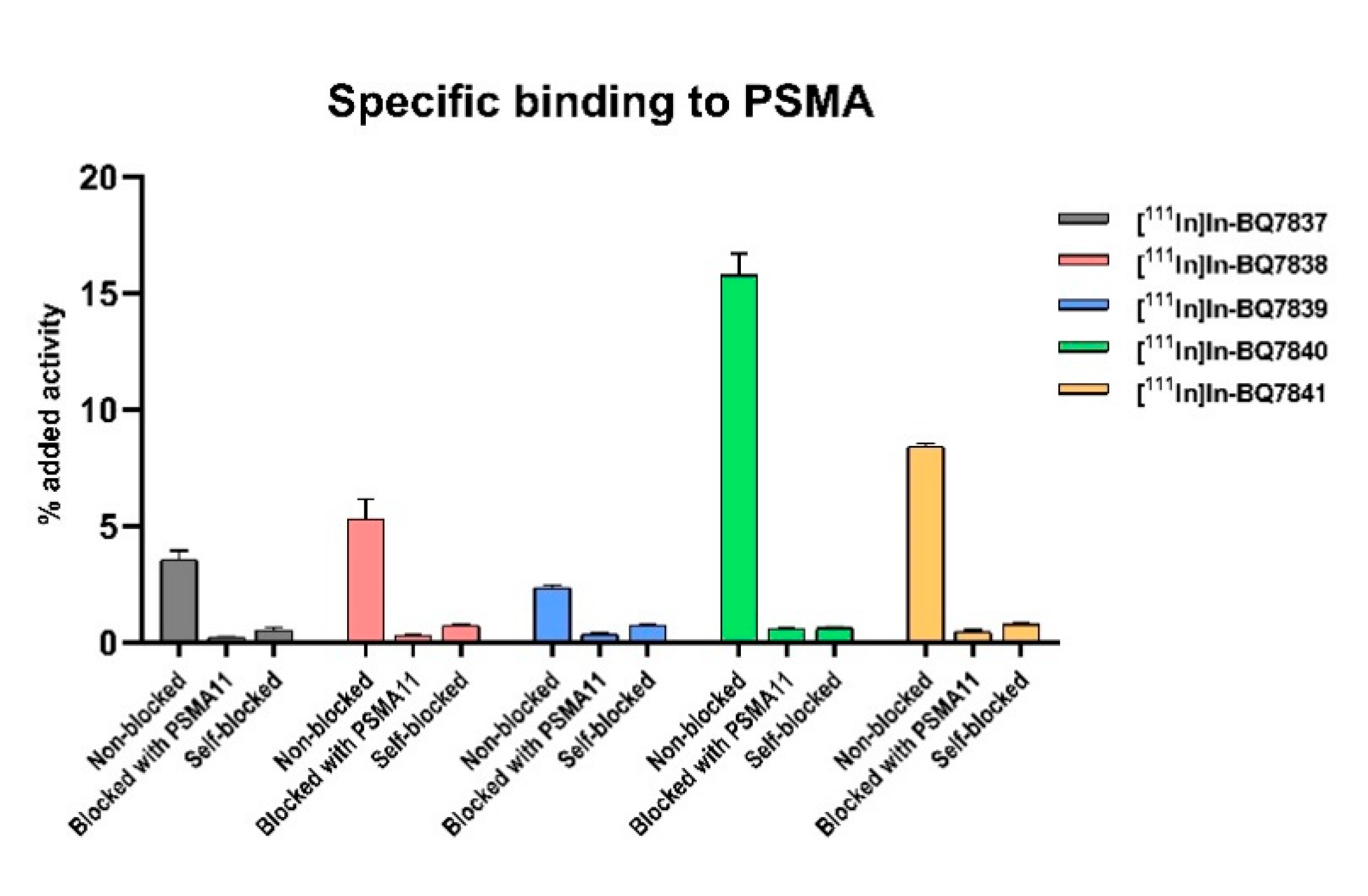
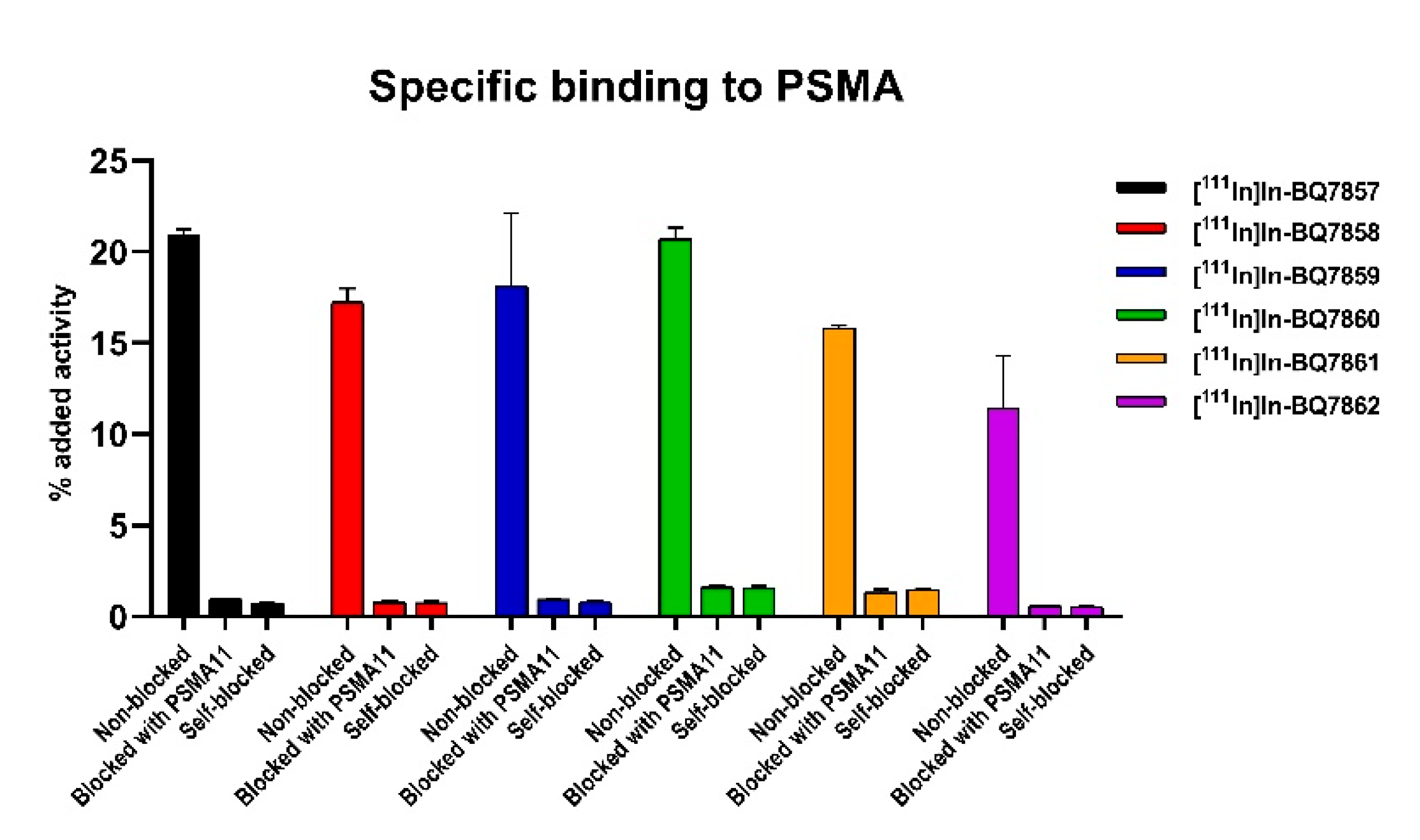
2.5.1. Specific Binding to PSMA
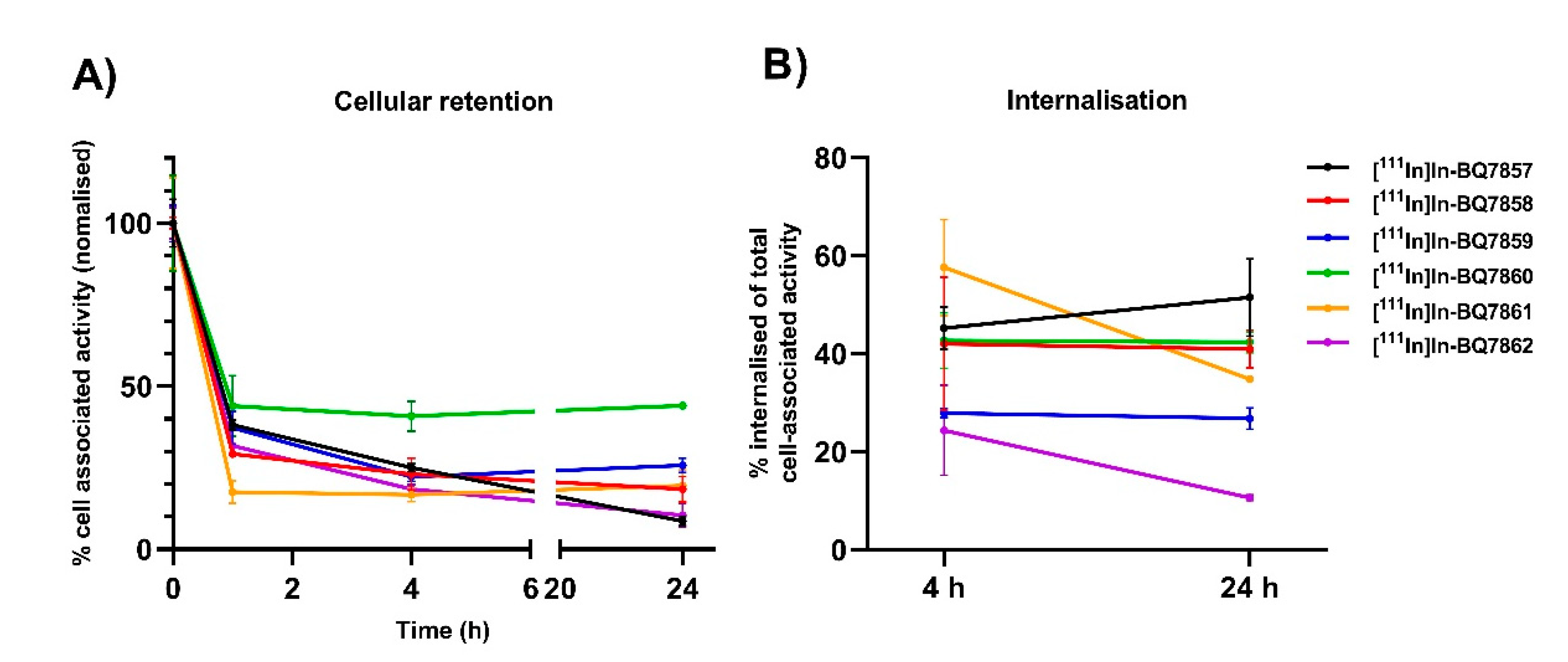
2.5.2. Cellular Retention
2.5.3. Real-Time Measurement of Binding Kinetics
2.6. In Vivo Characterisation
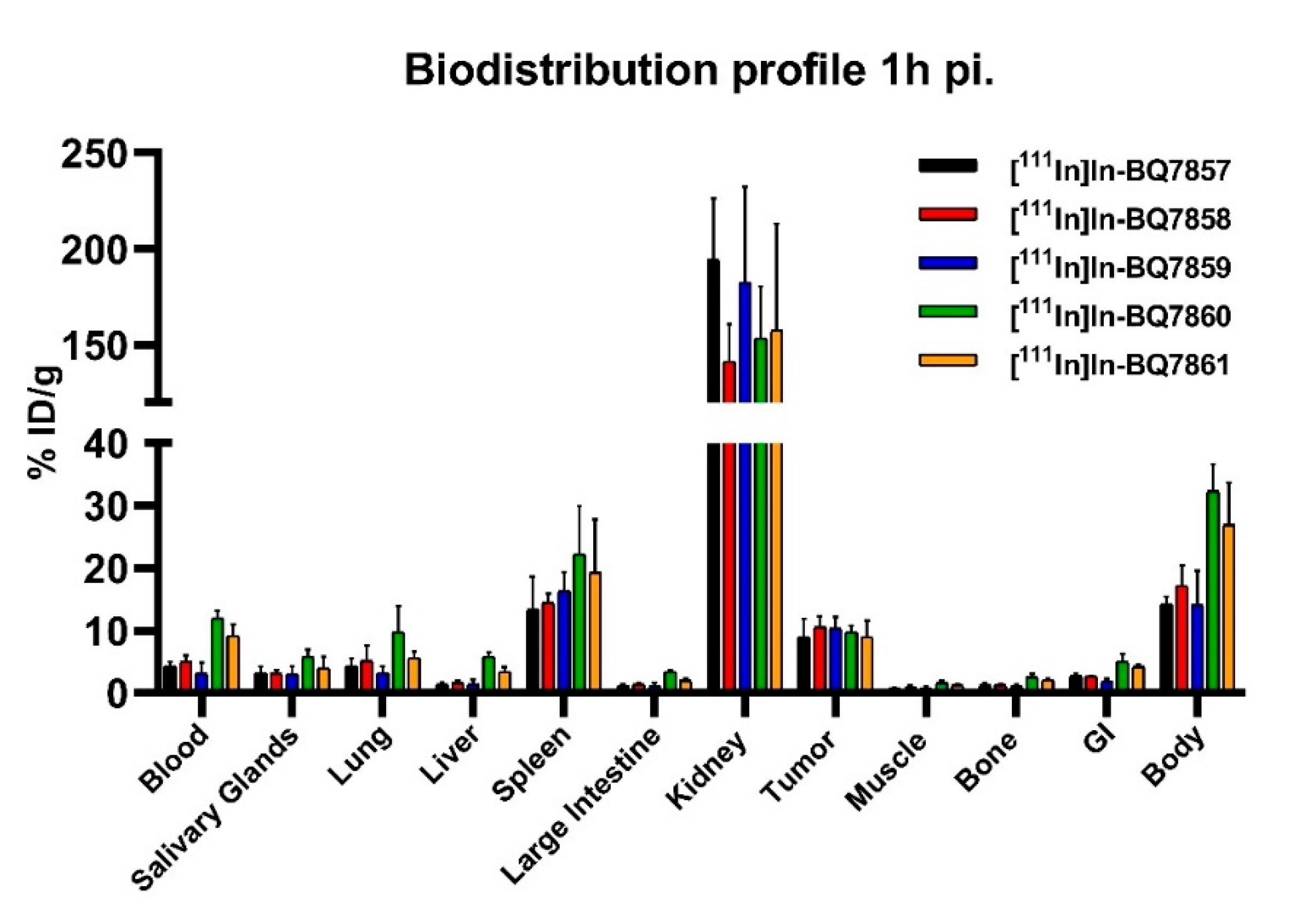
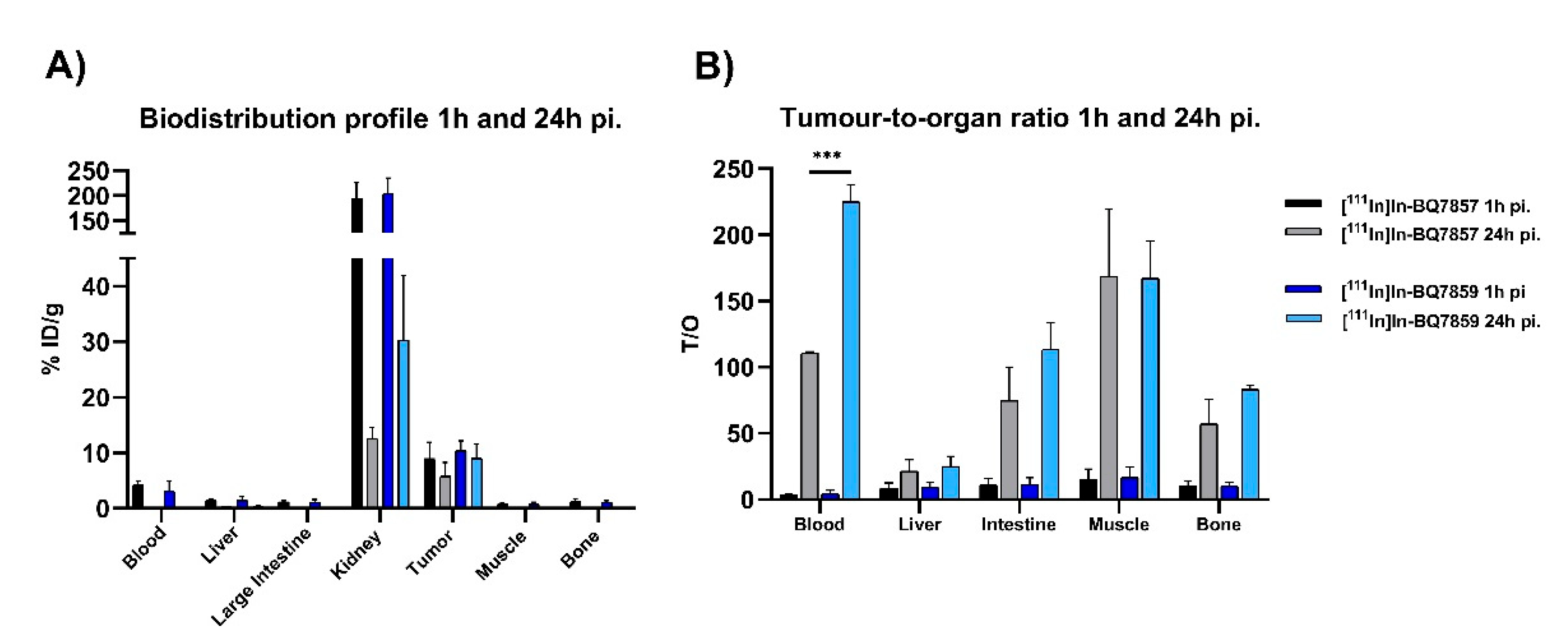
2.6.1. Biodistribution
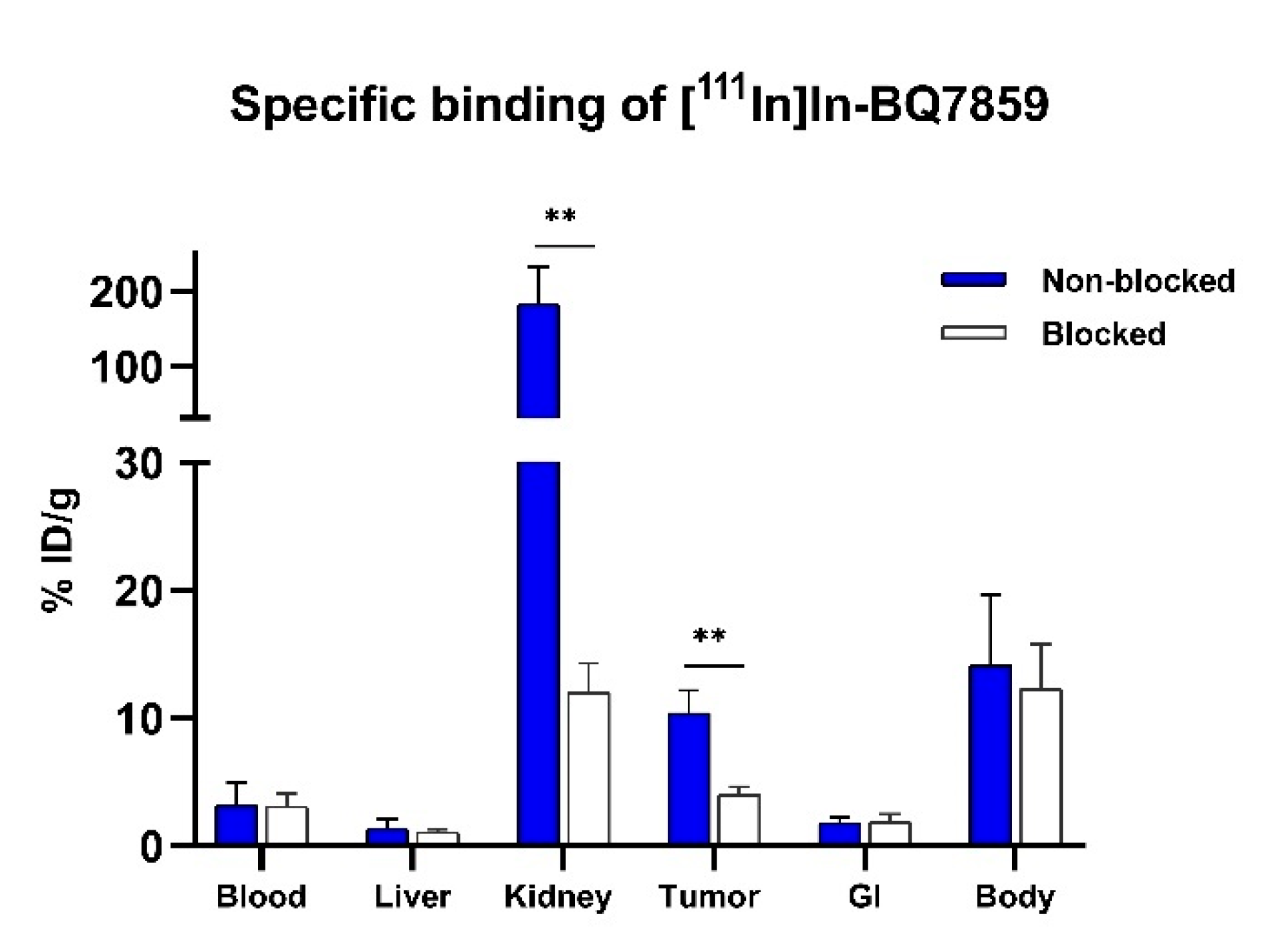
2.6.2. Specific Binding to PSMA
2.7. Statistical Analysis
3. Results and Discussion
3.1. Evaluation of BQ7837-41 (SET 1)
3.2. Evaluation of BQ7857-62 (SET 2)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. General Information
Appendix A.1.1. Instruments and Equipment
Appendix A.1.2. Chemicals and Solvents
Appendix A.1.3. Cell-lines
Appendix A.1.4. In Vivo model
References
- National Cancer Institute Surveillance, Epidemiology, and End Results Program Cancer Stat Facts: Prostate Cancer. Available online: https://seer.cancer.gov/statfacts/html/prost.html (accessed on 20 December 2021).
- Mottet, N.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer-2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Lavery, A.; Kirby, R.S.; Chowdhury, S. Prostate cancer. Medicine 2016, 44, 47–51. [Google Scholar] [CrossRef]
- Sarkar, S.; Das, S. A Review of Imaging Methods for Prostate Cancer Detection. Biomed. Eng. Comput. Biol. 2016, 7 (Suppl. 1), 1–15. [Google Scholar] [CrossRef] [Green Version]
- Histed, S.N.; Lindenberg, M.L.; Mena, E.; Turkbey, B.; Choyke, P.L.; Kurdziel, K.A. Review of functional/anatomical imaging in oncology. Nucl. Med. Commun. 2012, 33, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donin, N.M.; Reiter, R.E. Why Targeting PSMA Is a Game Changer in the Management of Prostate Cancer. J. Nucl. Med. 2017, 59, 177–182. [Google Scholar] [CrossRef]
- Barve, A.; Jin, W.; Cheng, K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J. Control. Release 2014, 187, 118–132. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.I.; Bennett, M.J.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef] [Green Version]
- Barinka, C.; Hlouchova, K.; Rovenska, M.; Majer, P.; Dauter, M.; Hin, N.; Ko, Y.S.; Tsukamoto, T.; Slusher, B.S.; Konvalinka, J.; et al. Structural basis of interactions between human glutamate carboxypeptidase II and its substrate analogs. J. Mol. Biol. 2008, 376, 1438–1450. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, S.; Riondato, M.; Uccelli, L.; Giovacchini, G.; Giovannini, E.; Duce, V.; Ciarmiello, A. Toward the Discovery and Development of PSMA Targeted Inhibitors for Nuclear Medicine Applications. Curr. Radiopharm. 2020, 13, 63. [Google Scholar] [CrossRef]
- Pavlíek, J.; Ptáek, J.; Bainka, C. Glutamate Carboxypeptidase II: An Overview of Structural Studies and Their Importance for Structure-Based Drug Design and Deciphering the Reaction Mechanism of the Enzyme. Curr. Med. Chem. 2012, 19, 1300–1309. [Google Scholar] [CrossRef]
- Kopka, K.; Benešová, M.; Bařinka, C.; Haberkorn, U.; Babich, J. Glu-ureido-based inhibitors of prostate-specific membrane antigen: Lessons learned during the development of a novel class of low-molecular-weight theranostic radiotracers. J. Nucl. Med. 2017, 58, 17S–26S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, P.; Ginj, M.; Walter, M.A.; Chen, J.; Reubi, J.C.; Maecke, H.R. Influence of different spacers on the biological profile of a DOTA-somatostatin analogue. Bioconjug. Chem. 2007, 18, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.S. Thermodynamics of Multivalent Interactions: Influence of the Linker. Langmuir 2010, 26, 8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maung, J.; Mallari, J.P.; Girtsman, T.A.; Wu, L.Y.; Rowley, J.A.; Santiago, N.M.; Brunelle, A.N.; Berkman, C.E. Probing for a hydrophobic a binding register in prostate-specific membrane antigen with phenylalkylphosphonamidates. Bioorg. Med. Chem. 2004, 12, 4969–4979. [Google Scholar] [CrossRef]
- Wester, H.J.; Schottelius, M.; Scheidhauer, K.; Meisetschläger, G.; Herz, M.; Rau, F.C.; Reubi, J.C.; Schwaiger, M. PET imaging of somatostatin receptors: Design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 117–122. [Google Scholar] [CrossRef]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Hadaschik, B.A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Haufe, S.; et al. Pet imaging with a [68ga]gallium-labelled psma ligand for the diagnosis of prostate cancer: Biodistribution in humans and first evaluation of tumour lesions. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 486–495. [Google Scholar] [CrossRef]
- Kiess, A.P.; Banerjee, S.R.; Mease, R.C.; Rowe, S.P.; Rao, A.; Foss, C.A.; Chen, Y.; Yang, X.; Cho, S.Y.; Nimmagadda, S.; et al. Prostate-specific membrane antigen as a target for cancer imaging and therapy. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 241–268. [Google Scholar]
- Jones, W.; Griffiths, K.; Barata, P.C.; Paller, C.J. PSMA theranostics: Review of the current status of PSMA-targeted imaging and radioligand therapy. Cancers 2020, 12, 1367. [Google Scholar] [CrossRef]
- Debnath, S.; Zhou, N.; McLaughlin, M.; Rice, S.; Pillai, A.K.; Hao, G.; Sun, X. PSMA-Targeting Imaging and Theranostic Agents—Current Status and Future Perspective. Int. J. Mol. Sci. 2022, 23, 1158. [Google Scholar] [CrossRef] [PubMed]
- Benesová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical evaluation of a tailor-made DOTA-conjugated PSMA inhibitor with optimized linker moiety for imaging and endoradiotherapy of prostate cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novartis Novartis PluvictoTM Approved by FDA as First Targeted Radioligand Therapy for Treatment of Progressive, PSMA Positive Metastatic Castration-Resistant Prostate Cancer. Available online: https://www.novartis.com/news/media-releases/novartis-pluvictotm-approved-fda-first-targeted-radioligand-therapy-treatment-progressive-psma-positive-metastatic-castration-resistant-prostate-cancer (accessed on 6 April 2022).
- Lundmark, F.; Abouzayed, A.; Mitran, B.; Rinne, S.S.; Varasteh, Z.; Larhed, M.; Tolmachev, V.; Rosenström, U.; Orlova, A. Heterodimeric Radiotracer Targeting PSMA and GRPR for Imaging of Prostate Cancer-Optimization of the Affinity towards PSMA by Linker Modification in Murine Model. Pharmaceutics 2020, 12, 614. [Google Scholar] [CrossRef] [PubMed]
- Mier, W.; Zitzmann, S.; Krämer, S.; Reed, J.; Knapp, E.M.; Altmann, A.; Eisenhut, M.; Haberkorn, U. Influence of Chelate Conjugation on a Newly Identified Tumor-Targeting Peptide. J. Nucl. Med. 2007, 48, 1545–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansch, C.; Fujita, T.; Jaffé, H.; Hine, J. Physical Organic Chemistry. Am. Chem. Soc. 1963, 85, 191. [Google Scholar]
- Mitran, B.; Varasteh, Z.; Abouzayed, A.; Rinne, S.S.; Puuvuori, E.; De Rosa, M.; Larhed, M.; Tolmachev, V.; Orlova, A.; Rosenström, U. Bispecific GRPR-Antagonistic Anti-PSMA/GRPR Heterodimer for PET and SPECT Diagnostic Imaging of Prostate Cancer. Cancers 2019, 11, 1371. [Google Scholar] [CrossRef] [Green Version]
- Eckelman, W.C.; Kilbourn, M.R.; Mathis, C.A. Specific to nonspecific binding in radiopharmaceutical studies: It’s not so simple as it seems! Nucl. Med. Biol. 2009, 36, 235–237. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Stefanova, M.; Benesova, M.; Bronzel, M.; Afshar-Oromieh, A.; Mier, W.; Eder, M.; Kopka, K.; Haberkorn, U. PSMA-Targeted Radionuclide Therapy of Metastatic Castration-Resistant Prostate Cancer with 177Lu-Labeled PSMA-617. J. Nucl. Med. 2016, 57, 1170–1176. [Google Scholar] [CrossRef] [Green Version]
- Mannweiler, S.; Amersdorfer, P.; Trajanoski, S.; Terrett, J.A.; King, D.; Mehes, G. Heterogeneity of prostate-specific membrane antigen (PSMA) expression in prostate carcinoma with distant metastasis. Pathol. Oncol. Res. 2009, 15, 167–172. [Google Scholar] [CrossRef]
- Minamimoto, R.; Hancock, S.; Schneider, B.; Chin, F.T.; Jamali, M.; Loening, A.; Vasanawala, S.; Gambhir, S.S.; Iagaru, A. Pilot comparison of 68Ga-RM2 PET and 68Ga-PSMA-11 PET in patients with biochemically recurrent prostate cancer. J. Nucl. Med. 2016, 57, 557–562. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Radiochemical Yield (%) | Release after 3 h (%) * | KD (nM) | |
|---|---|---|---|---|
| SET 1 | [111In]In-BQ7837 | 98.7 ± 0.4 | 3.1 ± 0.3 | 3130 |
| [111In]In-BQ7838 | 99.0 ± 0.3 | 1.6 ± 0.2 | 7520 | |
| [111In]In-BQ7839 | 99.0 ± 0.4 | 1.5 ± 0.5 | 7500 | |
| [111In]In-BQ7840 | 98.7 ± 0.4 | 3.4 ± 0.2 | 4.39 | |
| [111In]In-BQ7841 | 98.9 ± 0.4 | 1.5 ± 0.2 | 147 |
| Compound | Radiochemical Yield (%) | Release after 3 h (%) * | KD (nM) | LogD | |
|---|---|---|---|---|---|
| SET 2 | [111In]In-BQ7857 | 98.8 ± 0.5 | 5.6 ± 1.1 | 2.67 ± 0.4 | −2.97 |
| [111In]In-BQ7858 | 98.4 ± 0.4 | 7.0 ± 0.4 | 9.46 ± 1.3 | −2.45 | |
| [111In]In-BQ7859 | 99.1 ± 0.4 | 3.1 ± 0.6 | 6.53 ± 0.9 | −3.03 | |
| [111In]In-BQ7860 | 99.4 ± 0.4 | 3.6 ± 0.8 | 16.5 ± 4.6 | −1.42 | |
| [111In]In-BQ7861 | 98.4 ± 0.6 | 2.8 ± 0.3 | 12.4 ± 5.0 | −1.59 | |
| [111In]In-BQ7862 | 99.3 ± 0.3 | 1.3 ± 0.2 | 7.04 ± 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundmark, F.; Olanders, G.; Rinne, S.S.; Abouzayed, A.; Orlova, A.; Rosenström, U. Design, Synthesis, and Evaluation of Linker-Optimised PSMA-Targeting Radioligands. Pharmaceutics 2022, 14, 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14051098
Lundmark F, Olanders G, Rinne SS, Abouzayed A, Orlova A, Rosenström U. Design, Synthesis, and Evaluation of Linker-Optimised PSMA-Targeting Radioligands. Pharmaceutics. 2022; 14(5):1098. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14051098
Chicago/Turabian StyleLundmark, Fanny, Gustav Olanders, Sara Sophie Rinne, Ayman Abouzayed, Anna Orlova, and Ulrika Rosenström. 2022. "Design, Synthesis, and Evaluation of Linker-Optimised PSMA-Targeting Radioligands" Pharmaceutics 14, no. 5: 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14051098