Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers


School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China
*
Author to whom correspondence should be addressed.
Polymers 2020, 12(5), 1199; https://0-doi-org.brum.beds.ac.uk/10.3390/polym12051199
Submission received: 26 March 2020
/
Revised: 24 April 2020
/
Accepted: 28 April 2020
/
Published: 25 May 2020
(This article belongs to the Special Issue Polymer Matrix Composites for Advanced Applications II)
Abstract
:Alkynyl-terminated polyethylene oxide−tetrahydrofuran (ATPET) and glycidyl azide polymer (GAP) could be linked through click-chemistry between the alkynyl and azide, and the product may serve a binder for solid propellants. The effects of the energetic plasticizers A3 [1:1 mixture of bis-(2,2-dinitropropy) acetal (BDNPA) and bis-(2,2-dinitropropyl) formal(BDNPN)] and Bu-NENA [N-butyl-N-(2nitroxyethyl) nitramine] on the curing reaction between ATPET and GAP have been studied. A diffusion-ordered nuclear magnetic resonance spectroscopy (DOSY-NMR) approach has been used to monitor the change in the diffusion coefficient of cross-linked polytriazole polyethylene oxide−tetrahydrofuran (PTPET). The change in the diffusion coefficient of PTPET with A3 plasticizer is significantly higher than that of PTPET with Bu-NENA. Viscosity analysis further highlighted the difference between A3 and Bu-NENA in the curing process—the curing curve of PTPET (A3) with time can be divided into two stages, with an inflection point being observed on the fourth day. For PTPET (Bu-NENA), in contrast, only one stage is seen. The above methods, together with gel permeation chromatography (GPC) analysis, revealed distinct effects of A3 and Bu-NENA on the curing process of PTPET. X-ray Photoelectron Spectroscopy (XPS) analysis showed that Bu-NENA has little effect on the valence oxidation of copper in the catalyst. Thermogravimetric (TG) analysis indicated that Bu-NENA helps to improve the thermal stability of the catalyst. After analysis of several possible factors by means of XPS, modeling with Material Studio and TG, the formation of molecular cages between Bu-NENA and copper is considered to be the reason for the above differences. In this article, GAP (Mn = 4000 g/mol) was used to replace GAP (Mn = 427 g/mol) to successfully synthesize the PTPET elastomer with Bu-NENA plasticizer. Mechanical data measured for the PTPET (Bu-NENA) sample included ε = 34.26 ± 2.98%, and σ = 0.198 ± 0.015 MPa.
1. Introduction
Nowadays, the most widely used types of propellants include double-based propellant, butyl-hydroxyl propellant, NEPE propellant [NitrateEster Plasticized Polyether (NEPE) Propellant], and so forth. A3 [a 1:1mixture of bis-(2,2-dinitropropy) acetal (BDNPA) and bis-(2,2-dinitropropyl) formal (BDNPF)] and Bu-NENA [N-butyl-N-(2nitroxyethyl) nitramine] are two commonly used plasticizers in composite solid propellants. [1,2,3] These two plasticizers are widely applied in double-based and gun propellants, significantly improving their energy characteristics. [2,4] Recently, higher requirements have been placed on the performance indicators of solid composite propellants. It is hoped that they can be applied to NEPE propellants in order to further increase their energetic potential.
Click chemistry is a kind of classical chemical reaction between the azide and alkynyl groups and has been widely exploited since its discovery by Sharpless in 2001. [5] Its advantages include no side reaction, rapidity, high efficiency and no reaction with water, and so on. [6,7] This reaction has already been successfully applied in many fields, for example, biomedicine, polymer synthesis, [8] solid phase reaction, [9] and so forth. [10,11] Polytriazole polyethylene oxide−tetrahydrofuran (PTPET) elastomer is an example of a propellant successfully assembled by click chemistry in. [12,13] This elastomer is prepared from alkyne-terminated polyethylene oxide tetrahydrofuran co-polyether (ATPET) and glycidyl azide polymer (GAP). It has the potential to be used in NEPE propellants. However, an issue arises when A3 and Bu-NENA plasticizers are applied in PTPET-based propellant. Compared to those with A3, elastomers and propellants including Bu-NENA cannot be successfully cured into finished products. This phenomenon indicates that the curing system of PTPET including Bu-NENA plasticizer [PTPET (Bu-NENA), same as below] still has some problems, which limit its application in propellants. No published data on this issue are yet available. It is important to understand the role of the Bu-NENA in the curing process of the PTPET elastomer.
Calorimetry analysis is a classical method that can be used to evaluate the curing behavior of thermosetting elastomers. [14,15] In this study, it was necessary to analyze the effects of two plasticizers on the curing performance. The amounts of plasticizer used in the experiments were large, and their physical properties were quite different. Hence, it was impossible to obtain reliable information on curing of the elastomer by calorimetric analysis. However, Diffusion-ordered nuclear magnetic resonance spectroscopy (DOSY-NMR) has become a powerful tool for studying molecular interactions in solutions and determining molecular size. [16,17,18] It has also received increasing attention from chemical researchers, especially for exploring the interactions between subject and object, [19,20] molecular aggregation and other fields. [21] Diffusion is an inherent property of all molecules and varies with the size, shape, solubility, charge distribution, and so forth, of the particles. The nature of a mixture can also be analyzed by DOSY-NMR based on their diffusion coefficient of the proton environment.
In this article, we aim to confirm the effect of Bu-NENA on the curing of terminal alkynyl polytetrahydrofuran ethylene oxide copolyether and GAP by means of DOSY-NMR, viscosity and GPC analytical methods. Meanwhile, we discuss the potential effect of the presence of the Bu-NENA in PTPET. It is hoped that the collected data may be helpful in solving the above mentioned problems by comparative analysis of experimental data and the results of theoretical calculations for the systems with A3 and Bu-NENA.
2. Experimental
2.1. Materials and Methods
ATPET (Mn = 4000 g/mol) was synthesized in our laboratory. [22] GAP (Mn = 427 g/mol, the average functionality of −N3 was 3.8) and GAP (Mn = 4000 g/mol, the average functionality of −N3 was 38) was used as the curing agent and offered by Liming Chemical Research Institute. A3 and Bu-NENA were the two plasticizers used in this experiment provided by Xi’an North Huian Chemical Industry Co., Ltd. (Xi’an, China). The mass ratio of plasticizer to ATPET in this experiment is 1:1. A mole ratio of 1:1 for the azide groups to the alkynyl groups was obtained by using GAP (Mn = 427 g/mol), [23] whereas 10:1 was achieved by using GAP (Mn = 4000 g/mol) experiment. The PTPET elastomer was synthesized by the click-chemistry reaction between ATPET and GAP (Mn = 427 g/mol) in the presence of a copper catalyst at 50 °C for 7 days. A schematic diagram of the synthesis method for the PTPET elastomer is shown in Scheme 1 [13].
2.2. Measurements and Analysis
Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker Avance HD 600 instrument (Bruker, Karlsruhe, Germany) at 299.15 K. The ledbpgp2s1d spin echo sequence was applied for DOYS-NMR experiments. Sixteen points were recorded to produce each 1H-NMR spectrum. The delay between experiments (d1) was set to 1 s. (D16: 0.0002 s, D20: 0.5 s, D21: 0.005 s, GPZ7: −17.13%, GPZ8: −13.17%, P100: 600, P30: 1000). Each prepared sample (100 mg) was added to a 5 mm NMR tubes containing 0.6 mL of CDCl3. The viscosity of each reaction system was measured with a HAAKE MARS iQ apparatus (Thermo Fisher Scientific, Waltham, MA, USA). Molecular weights and their distributions were determined by gel permeation chromatography (GPC, Waters-1515, Atlanta, GA, USA). Thermogravimetric analysis (TGA) was performed on a Netzsch 209 F1 instrument (Selb, Germany, Al2O3 crucible) instrument with the following test conditions—test atmosphere N2, scanning rate 10 °C/min and temperature range 40~500 °C. X-ray photoelectron spectroscopy (XPS) was performed on a PHI QUANTERA-II SXM (ULVAC-PHI, Kanagawa, Japan) apparatus using the C1s peak (B.E. = 284.6 eV) as a reference.
Molecular dynamics (MD) simulation analysis of the compatibility and interaction between the plasticizers A3/Bu-NENA and polymer was performed using the amorphous builder and forcite modules in Material Studio (MS) 6.0 software. Molecular models of ATPET binder with Bu-NENA, and A3 (composed of BDNPF and BDNPA mixed in equal mass ratio) were established according to their respective structural formulae (as shown in Figure 1) by MS. The mass ratio of ATPET to plasticizer in the model is 1:1. The detailed experimental parameters of MS simulation were based on using the reference parameters. [24] (Forcefield: Compass II, Buffer width: 0.5 Å, Cutoff distance: 12.5 Å).
3. Results and Discussion
3.1. H-NMR and DOSY Analysis
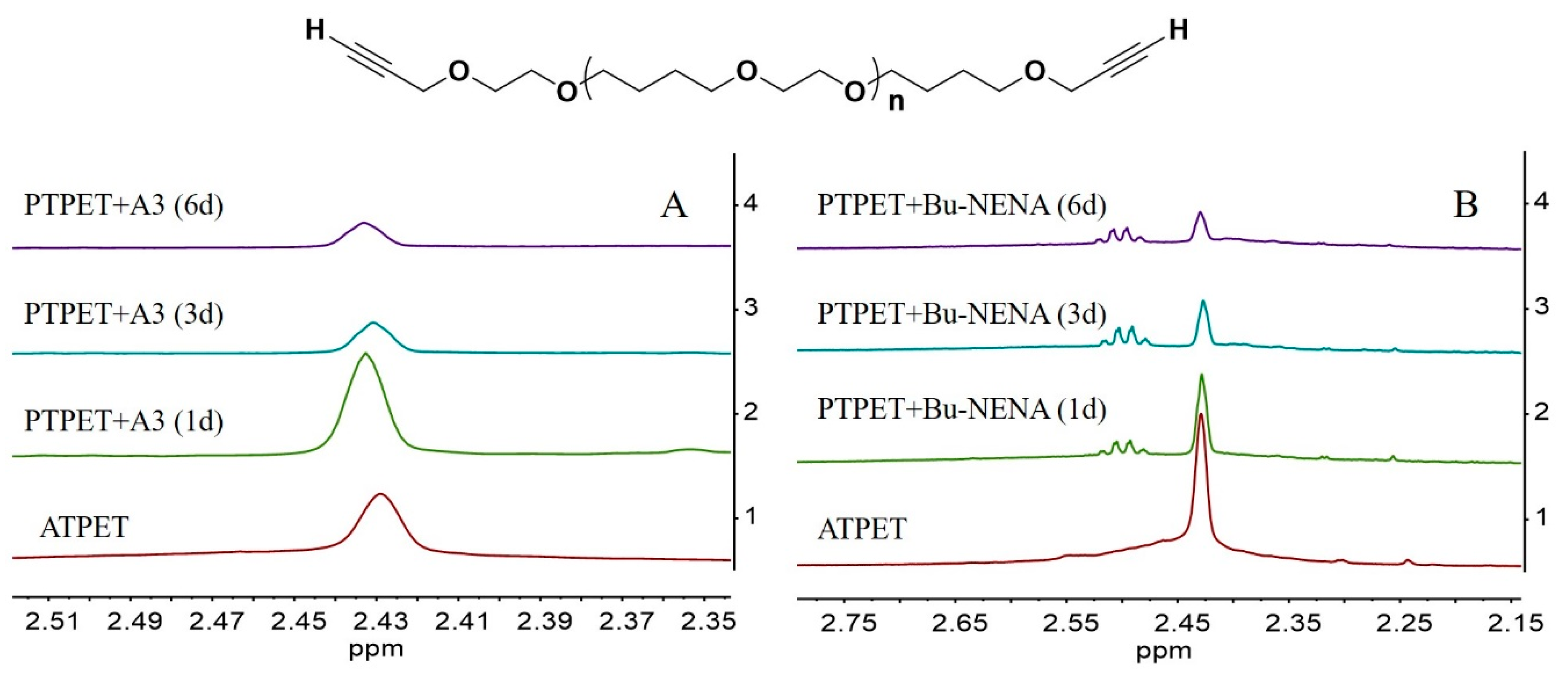
Extensive peak overlap precluded the identification of single components in the mixtures by 1H-NMR. However, some information could still be gleaned from non-overlapping peaks. The peak at 2.45 ppm can be ascribed to the signal of the hydrogen atom on the terminal alkynyl group in ATPET. The changes in the 1H-NMR spectra with time for the reaction systems with plasticizers A3 and Bu-NENA are shown in Figure 2. It is clear that for both plasticizer systems, the intensity of this peak decreases with time. This phenomenon is consistent with partial consumption of the terminal alkynyl groups through click reaction with the azide groups in ATPET.
Clearly, it was difficult to unequivocally identify and quantify the individual components in this complex system by 1H-NMR. [25] To get more information, high-field nuclear magnetic resonance (NMR) diffusion equipment was used to monitor the phase transition molecular dynamics during polymer formation occurring in the liquid systems containing the respective plasticizer. DOSY-NMR is an effective approach for separating overlapping peaks by plotting chemical shift against the diffusion coefficient. In previous literature, an empirical formula between diffusion coefficient and molecular weight has been established, allowing different substances to be distinguished. [26] The DOSY-NMR method has proven to be very valuable for identifying components in unknown samples.
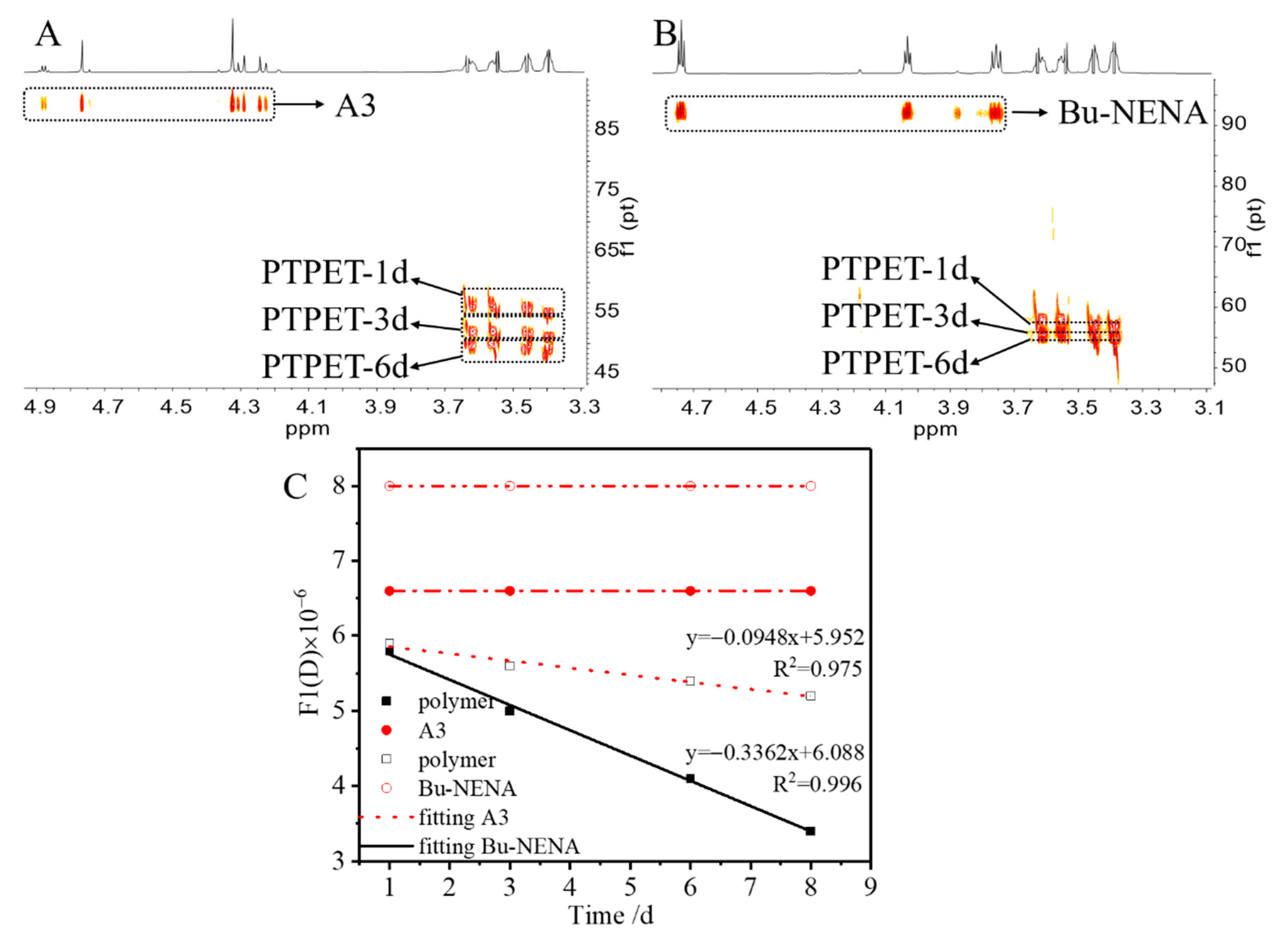
DOSY spectra of the plasticizers and polymer mixtures after different times are shown in Figure 3; (A) A3 and polymer; (B) Bu-NENA and polymer. All samples were tested at the same concentration, with the same test parameters and NMR pulse sequence. The experimental results of different test time were superimposed on an analysis. The DOSY spectra indicated good separation of the plasticizer and polymer in the reaction solution. The peaks in the dashed boxes showed very similar diffusion coefficients, implying that they belonged to the same compound. The signals in the upper dashed boxes in Figure 3A,B are those of the plasticizers A3 and Bu-NENA, respectively, and their diffusion coefficients do not change with time. They have larger diffusion coefficients than that of the polymer. The signals in the bottom right-hand corners of Figure 3A,B are those of the PTPET polymer. By comparing and analyzing these results, it can clearly be seen that the degrees of polymerization in the presence of the respective plasticizers are significantly different, in accordance with the correlation between the molecular weight of a polymer and its diffusion coefficient. [27] The diffusion coefficient of the polymer in the A3 system is significantly larger than that in the Bu-NENA, indicating that the polymer produced in the former system is much larger. In order to further understand the differences between the respective systems, the relationship between diffusion coefficient and time or each substance was analyzed, and the results are shown in Figure 3C. The relationship between the diffusion coefficient of the polymer and time in the A3 system can be expressed as y = −0.0948x + 5.952, with R2 = 0.975, whereas that in the Bu-NENA system can be expressed as y = −0.3362x + 6.088, with R2 = 0.996.
3.2. Viscosity Analysis
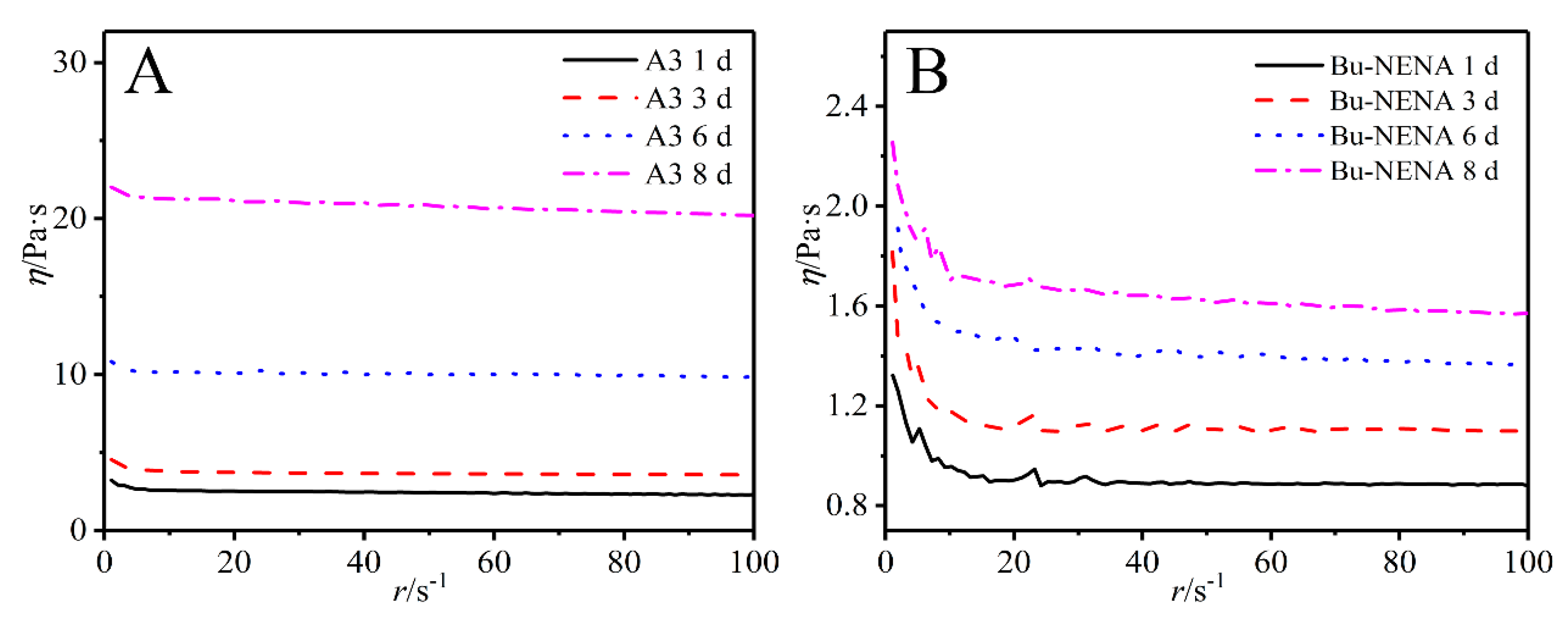
The intrinsic viscosities of samples of the A3 and Bu-NENA systems were determined by stress rheometer measurements in the range 1–100 rad/s. The main linkage of the s molecular chain of the ATPET is the C-O bond. Consequently, the internal rotation barrier is small, the molecular chain is flexible, and the fluidity of the material is also good. Viscosity change curves with respect to time are shown in Figure 4, for the A3 (A) and Bu-NENEA (B) systems. With increasing reaction time, the molecular weight of the PTPET polymer gradually increases due to the click chemistry reaction between ATPET and GAP. It is well known that the viscosity of a polymer is a function of molecular weight, shear conditions, temperature, and pressure [28]. Curves A and B in Figure 4 verify that the viscosities of both polymer systems gradually increased with reaction time. However, distinct trends can be discerned for the respective systems. The viscosity increase of the A3 system was significantly greater than that of the Bu-NENA system.
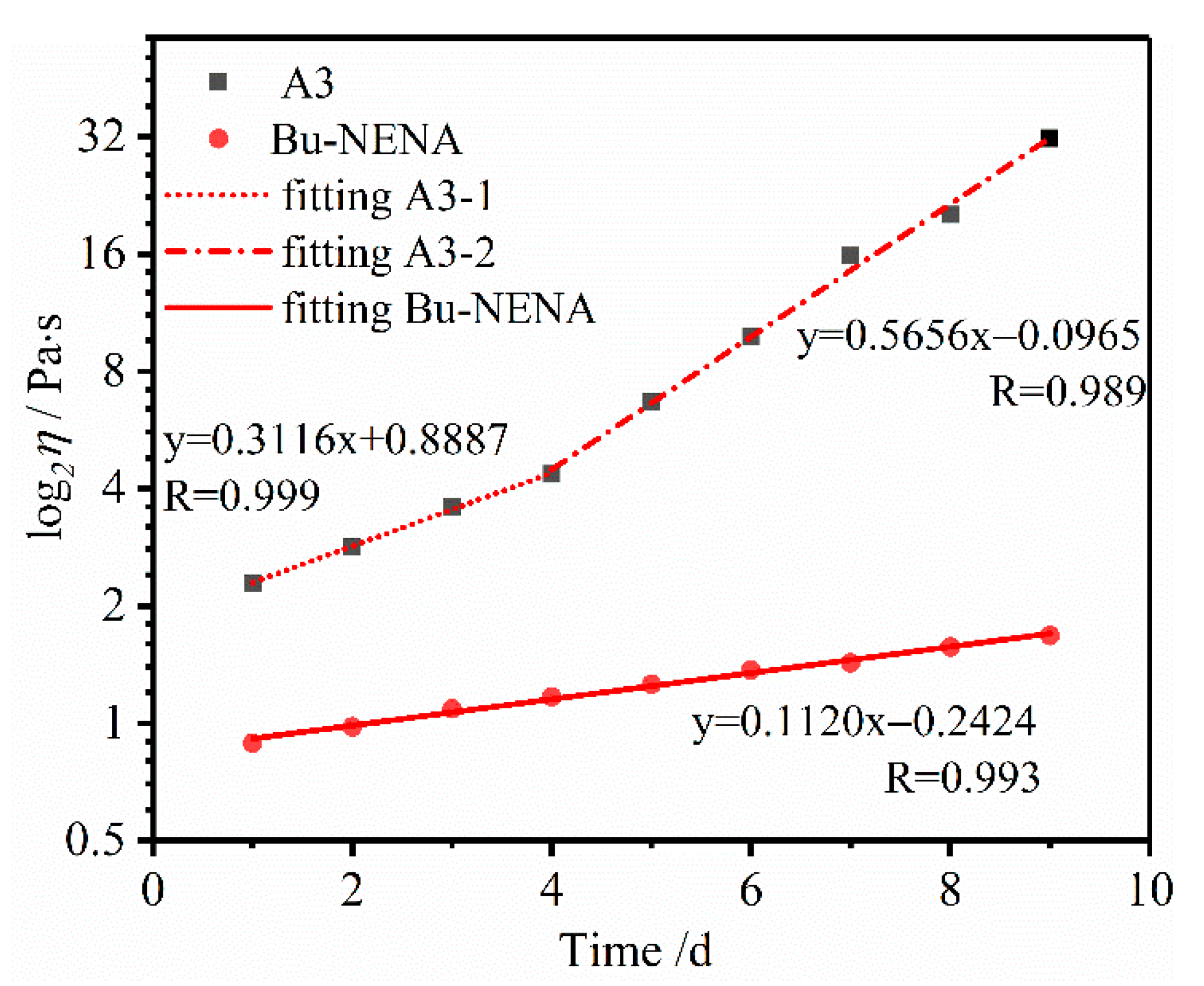
In order to further analyze the change trends of the viscosity in the two systems, the data were subjected to statistical analysis, and depicted in Figure 4. The collected data points were utilized to fit a plot of log2η vs. time. Results for the Bu-NENA system show a good linear regression; the fitting function is y = 0.1120x − 0.2424, with R = 0.993. However, there are obvious differences for the A3 system. The first half of the data shows as a good fitting linear relationship, for which the fitting function is y = 0.3116x + 0.8887, with R = 0.999. The second half of the data shows another good linear fitting relationship, the fitting function for which is y = 0.5656x − 0.0956, with R = 0.989.
According to the theory of the polymer material flow, molecular weight is one of the main factors affecting the rheological properties of polymers. [29] The relationship between the molecular weight and viscosity is expressed by the simple equations:
where Mc is the critical molecular weight of the polymer, which is related to the molecular weight restricted by the entanglement of polymer segments.
Once the molecular weight exceeds Mc, mutual entanglement occurs between the molecular chains. Interactions between the molecular chains suddenly increase due to this entanglement, and the stress on one molecular chain will be transferred to other molecular chains, significantly increasing the viscosity of the polymer. As shown in Figure 5, an inflection point in the viscosity curve of the A3 system is seen after 4 days. This experimental phenomenon implies that the molecular weight of the polymer in A3 could exceed the gel point. This is an important factor with regard to the application of A3 and Bu-NENA plasticizers in PTPET-based propellant. A3 elastomer and propellants can be successfully cured into final products, but systems with Bu-NENA cannot.
3.3. Gel Permeation Chromatography (GPC) Analysis
Gel permeation chromatography (GPC) is an effective technique for characterizing the molecular weight changes of polymers. In order to further corroborate the validity of the viscosity analysis, the molecular weight changes of the polymer in the A3- and Bu-NENA-containing systems were tracked, and the results are presented in Figure 6. 10 mg of the polymer sample and 0.5 mL of THF were added to a centrifuge tube. After the sample is fully dissolved, the mixture was filtered with a 0.2 μm filter membranes and take the filtrate for analysis and testing. Thermosetting elastomers can be formed by linear polymers through cross-linking reactions. Therefore, the molecular weight of the uncrosslinked portion will gradually increase with the increase of reaction time and reaction degree, and the polydispersity index (PDI) will gradually become wider. The GPC analysis results for the two systems show that the peak time of the polymer shifts to the left with increasing reaction time. From Figure 6A,B, it can be seen that there was a significant difference in the molecular weights of the polymers in the respective systems. The amplitude of the left shift of the curve for the A3 system is significantly higher than that for the Bu-NENA system, and the polydispersity index (PDI) is also wider for the former than for the latter. The Mn and PDI analysis data of the studied samples are shown in Table 1. Although the GPC data obtained for the polymers related to the uncrosslinked part of the system, the results are still somewhat enlightening. GPC analysis data are consistent with the viscosity and DOSY NMR analysis described above.
Through the above experimental analysis by NMR, GPC and viscosity, it is confirmed that there are indeed differences between the A3- and Bu-NENA-containing systems. In order to further delineate the reasons for the differences between the above two systems, the following sections outine several possibilities and gives the corresponding analysis and characterization data.
3.4. X-ray Photoelectron Spectroscopy Analysis
One possibility is that Bu-NENA inactivates the catalyst and oxidizes monovalent copper to divalent copper, which may cause failure of the click chemical reaction. In order to test this hypothesis, a catalyst sample, 10 mg was immersed in Bu-NENA (1 mL). The mixture was placed in an oven at 50 °C for 5 d, then vacuum-dried and sampled for XPS analysis. XPS results for the pure catalyst and the Bu-NENA soaked catalyst are displayed in Figure 7. Only two peaks were seen in the XPS analysis of the pure catalyst, namely the 2p 1/2 peak of Cu+ in the catalyst at 952.3 eV and the 2p 3/2 peak at 932.4 eV. [30] The catalyst treated with Bu-NENA showed a small peak at 934.9 eV attributable to Cu2+, and a small satellite peak at 944.0 eV. However, the signal intensities of these two peaks were only slightly higher than the signal-to-noise ratio. This observation indicated that only a small fraction of the Cu+ in the catalyst soaked with Bu-NENA was oxidized to Cu2+. It could be observed from the peak areas that most of the Cu+ had not been oxidized and was still monovalent. The samples treated with Bu-NENA and the original sample both showed the green color of monovalent copper. The above analysis shows that oxidation of the catalyst from Cu+ to Cu2+ by Bu-NENA was not the main reason for suppression of the reaction.
3.5. Solubility Parameter Analysis
Miscibility refers to the property of a polymer and plasticizer to form a macroscopically homogeneous material. The parameter of cohesive energy density (CED) has a direct effect on the physical characteristics of the polymer. The solubility parameter (δ) plays an important role in the theory of mixtures. [31] There is also speculation that the compatibility of ATPET with Bu-NENA and A3 plasticizers is different, making the reactant system unable to mix uniformly, resulting in the observed reactivity difference.
The CED and δ parameters of the two systems were calculated using molecular dynamics, and the results are shown in Table 2. The CED values of Bu-NENA and A3 are both over 420 J cm−3, classifying them as polar plasticizers. The ATPET matrix, meanwhile, is a low-polarity polymer, since its CED is 338.9 J cm−3. In our study, it is clear that the magnitudes of Δδ between the two polymers and plasticizer systems are < 2.1 (J cm−3)0.5. Based on the miscibility principle of similar material structures, when δ of the additive is close to that of the polymer, they can dissolve well in one another. Therefore, the probability of this explanation is low.
CED = CEDv + CEDe
δ = δv + δe.
However, this approach is not suitable for systems with strong hydrogen bonds between the components. This is because the theory of solubility parameters only considers the influence of intermolecular dispersion forces, and ignores dipole forces and hydrogen-bonding interactions.
3.6. Radial Distribution Function Analysis
Intermolecular interactions are strongly influenced by steric effects. The radial distribution function (RDF) can be used to reveal the interactions of different atoms of ATPET with plasticizers.
The distance at which a peak appears an effective approach for inferring the type of interaction force. Generally, the van der Waals force is from 0.31 nm to 0.50 nm, while the distances of hydrogen bonds are from 0.26 nm to 0.31 nm. [32,33] The value of the force is judged by the peak height in the RDF curve.
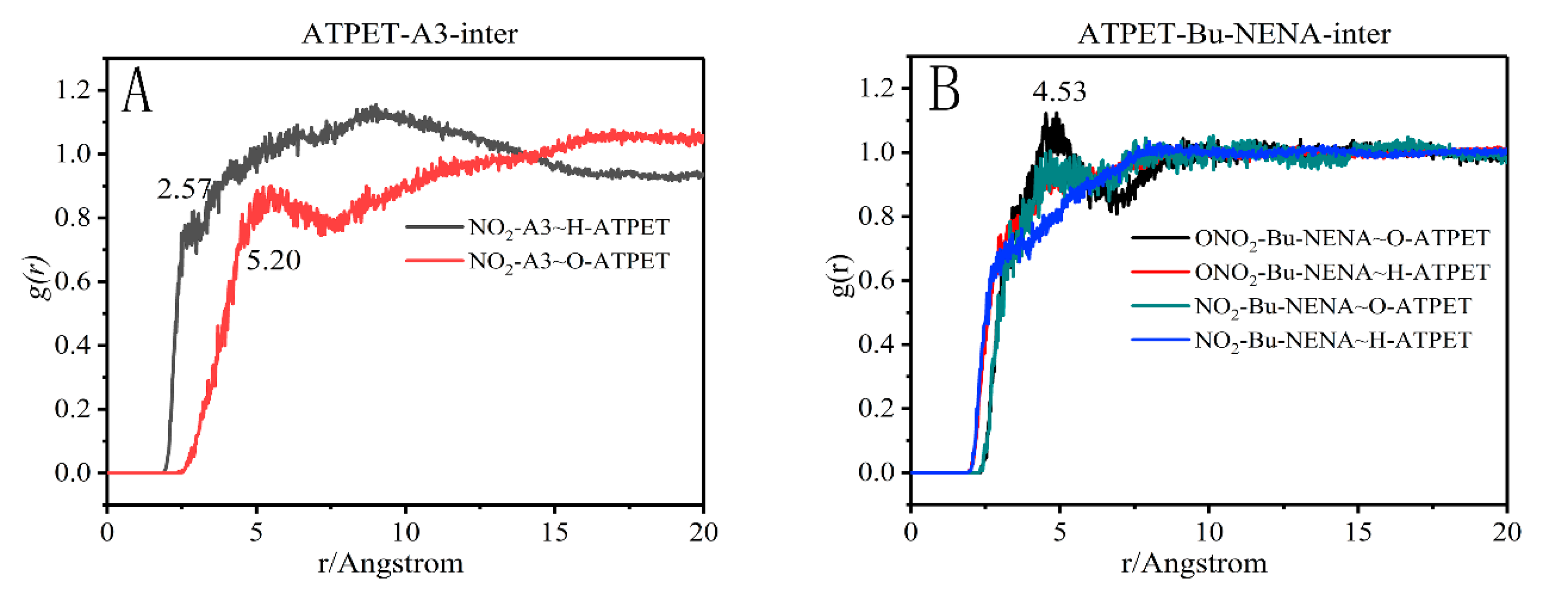
The RDF curves for interactions between specific moieties of ATPET and the plasticizers (A3/Bu-NENA) are shown in Figure 8. There is a peak at 2.57 Å which g(r) = 0.81 for the pair of H in ATPET and the NO2 group in A3; the peak for O in ATPET and the NO2 group in A3 is at 5.20 Å which g(r) = 0.89. In the RDF curves for the Bu-NENA system, there is a peak at 4.53 Å which g(r) = 1.00 for the pair of O in PTPET and the NO2 group in Bu-NENA; the peak for H in ATPET and the NO2 group in Bu-NENA is at 2.91 Å which g(r) = 0.69. Meanwhile, for the relationship between the ONO2 group and atoms in ATPET, there is a main peak at 4.53 Å which g(r) = 1.12 for the pair of O in ATPET and the ONO2 group in Bu-NENA; the peak for H in ATPET and the ONO2 group in Bu-NENA is at 3.35 Å which g(r) = 0.79. From comprehensive analysis of the RDF curves, the interaction forces of NO2-A3~H-ATPET and NO2-Bu-NENA~H-ATPET are hydrogen bonds, while the interaction forces of NO2-A3~O-ATPET, NO2-Bu-NENA~O-ATPET, ONO2-Bu-NENA~O-ATPET and ONO2-Bu-NENA~H-ATPET are van der Waals interactions. The highest peak probably for a strong interaction force is between the ONO2 group of Bu-NENA and the O in ATTPE. The value of g(r) for the Bu-NENA system sharply increased by about 2.5 Å, indicating that the effect of hydrogen-bonding was greater than in the A3 system.
3.7. Thermogravimetric (TG) Analysis
According to the above experimental and computational analyses, it is clear that the above three possibilities are not the main factors that prevent PTPET from curing. From previous literature on the CuAAC reaction, a specific phenomenon is that copper complexes are more susceptible to inhibition by forming molecular cages in some ligand environments. [34] Bu-NENA competes with the complexing ligand in the copper catalyst, inactivating the copper center and preventing the CuAAC reaction.
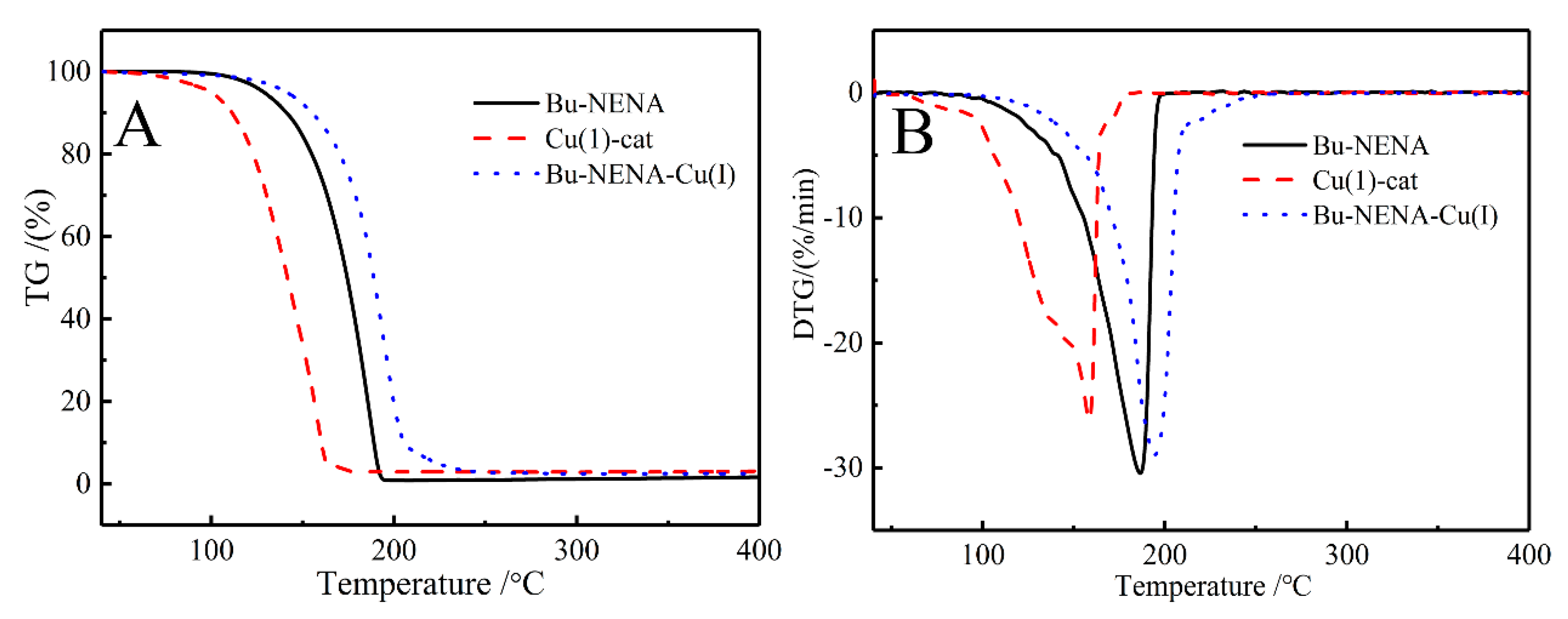
As the ONO2 group in Bu-NENA is electron-donating, it is speculated that Bu-NENA inhibits catalysis of the CuAAC reaction through the formation of an inhibitory molecular cage with copper. The catalyst (10 mg) was dissolved in Bu-NENA (1mL) to form a homogeneous solution. A sample was then subjected to thermogravimetric analysis, and the results are exhibited in Figure 9. Thermal decomposition onset temperatures (T95%) for the respective samples were as follows—Tcat-95% = 99.9 °C, TBu-NENA-95% = 128.9 °C, Tmix-95% = 141.2 °C. The maximum decomposition temperatures (Tm) were as follows—Tcat-m = 158.6 °C, TBu-NENA-m = 186.5 °C, Tmix-m = 194.5 °C. The values of T95% and Tm for the sample were greatly improved after mixing. The above experimental data give supplementary evidence that Bu-NENA forms a complex with copper in the catalyst and modifies its properties.
3.8. An Alternative Method
How to apply Bu-NENA in solid propellants is still an issue that urgently needs to be addressed because the energy of Bu-NENA is higher than that of A3. In view of the fact that the above-mentioned molecular cage structure cannot perform CuAAC reaction, thermal catalysis of the reaction between an azide and an alkynyl group to form a triazole ring has become a possible solution.
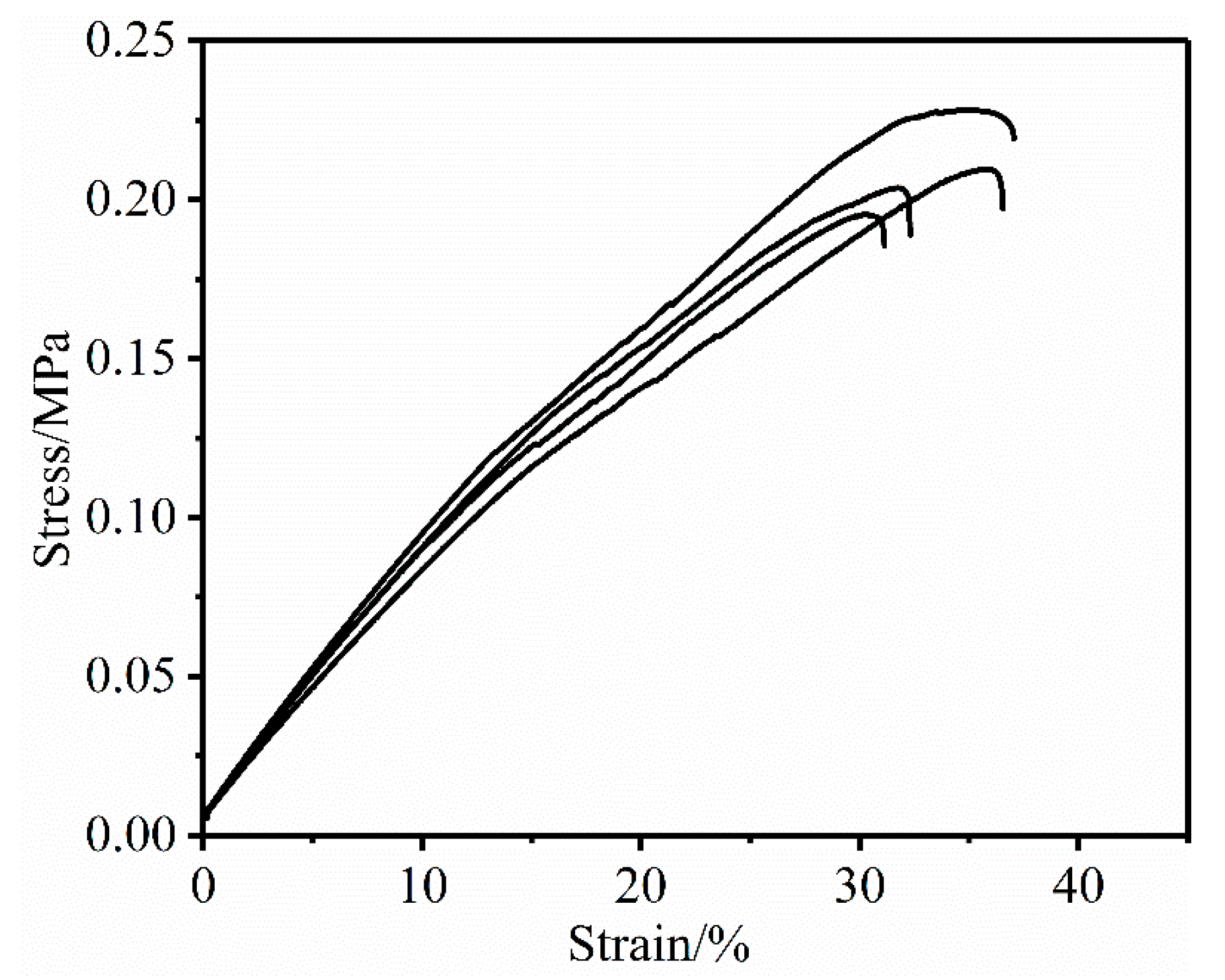
GAP (Mn = 4000 g/mol, R = 38) was used to replace GAP (Mn = 427 g/mol, R = 3.8) to form PTPET elastomer (including Bu-NENA) without catalyst. An PTPET (mATPET: mBu-NENA = 1:1) elastomer was successfully formed after 7 days at 50 °C, and its mechanical properties was shown in Figure 10: ε = 34.26 ± 2.98%, σ = 0.198 ± 0.015 MPa. However, there is still a big gap for the mechanical properties between this container Bu-NENA elastomer and the PTPET elastomer without Bu-NENA. For the PTPET elastomer—ε = 228.45%, σ = 0.69 MPa. [35] The above data is also much lower than the A3 plasticized Glycidyl Azide Polymer-Polycaprolactone-Polyurethane elastomer system, which has a tensile strength of 1.8 MPa and a tensile strain at break of 1780%. [36] This experiment showed by using GAP (Mn = 4000 g/mol), the PTPET elastomer could be cured in the presence of plasticizer Bu-NENA. Although the mechanical properties of PTPET (including Bu-NENA) elastomers are still relatively low, this still has great significance. This is an example report of the successful preparation of PTPET (including Bu-NENA) elastomers.
4. Conclusions
DOSY-NMR, viscosity and GPC results have confirmed that there is indeed a considerable difference between the plasticizers A3 and Bu-NENA during the curing of PTPET. The curing reaction between ATPET and GAP is less efficient in the presence of Bu-NENA, but the precise mechanism needs further research. Monovalent copper in the catalyst is oxidized to a divalent state. Bu-NENA is decomposed to generate NO2, accounting for some differences between the properities of Bu-NENA and A3. Several possibilities for the differences between the two plasticizers A3 and Bu-NENA have been proven not to be the main factors that prevent PTPET from curing. The formation of molecular cages between the catalyst and Bu-NENA seems to be the most probable reason for the difference, but the specific mechanism needs further in-depth study. Finally, we propose a method of applying thermal catalysis to increase reaction sites after comprehensive analysis of the curing of azide and alkynyl groups. After a replacement of GAP (Mn = 4000 g/mol) with GAP (Mn = 427 g/mol), the curing of PTPET elastomer containing Bu-NENA plasticizer was successfully achieved.
It is hoped that this article gives an important indicator for the application of PTPET elastomers in composite propellants. However, the mechanical properties of the prepared elastomer are still relatively poor and cannot meet current production needs. Improving of the mechanical properties will be a further focus of our future work.
Author Contributions
J.H. (Jinghui Hu) and J.H. (Jiyu He) directed the research, J.H. (Jiyu He) designed the experiments, J.H. (Jinghui Hu), Y.L., and K.C. performed the experiments, J.H. (Jinghui Hu) and J.H. (Jiyu He) analyzed the data and prepared the manuscript, and J.H. (Jiyu He) and R.Y. edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank the staff of the School of Materials Science and Engineering Beijing Institute of Technology for the measurement. We also thank Lan Yanhua of North University of China for her help in Material Studio calculation. The authors declare no competing financial interest.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Damse, R.S.; Singh, A. Evaluation of energetic plasticisers for solid gun propellant. Def. Sci. J. 2008, 58, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Damse, R.S.; Omprakash, B.; Tope, B.G.; Chakraborthy, T.K.; Singh, A. Study of N-n-butyl-N-(2-nitroxyethyl)nitramine in RDX based gun propellant. J. Hazard. Mater. 2009, 167, 1222–1225. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, J. Review on Synthesis of BDNPA/F Plasticizer and its Development. Chin. J. Energ. Mater. 2007, 1, 90–93. [Google Scholar]
- Zhao, B.; Xia, M.; Huang, J.; Li, W.; Liu, Q.; Luo, Y. Characterization of Bu-NENA/NC Low Sensitive Double-base Propellant. Chin. J. Energ. Mater. 2017, 25, 794–798. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004. [Google Scholar] [CrossRef]
- Demko, Z.P.; Sharpless, K.B. A click chemistry approach to tetrazoles by Huisgen 1,3-dipolar cycloaddition: Synthesis of 5-sulfonyl tetrazoles from azides and sulfonyl cyanides. Angew. Chem. Int. Ed. 2002, 41, 2110–2113. [Google Scholar] [CrossRef]
- Diaz, D.D.; Punna, S.; Holzer, P.; McPherson, A.K.; Sharpless, K.B.; Fokin, V.V.; Finn, M.G. Click chemistry in materials synthesis. 1. Adhesive polymers from copper-catalyzed azide-alkyne cycloaddition. J. Polym. Sci. Part A-Polym. Chem. 2004, 42, 4392–4403. [Google Scholar] [CrossRef]
- Hawker, C.J.; Fokin, V.V.; Finn, M.G.; Sharpless, K.B. Bringing efficiency to materials synthesis: The philosophy of click chemistry. Aust. J. Chem. 2007, 60, 381–383. [Google Scholar] [CrossRef]
- Castro, V.; Rodriguez, H.; Albericio, F. CuAAC: An efficient click chemistry reaction on solid phase. ACS Comb. Sci. 2016, 18, 1–14. [Google Scholar] [CrossRef]
- Meldal, M. Polymer “Clicking” by CuAAC reactions. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhai, J.; Yang, R. Comparison between properties of polyether polytriazole elastomers and polyether polyurethane elastomers. Polym. Adv. Technol. 2014, 25, 314–321. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Xiao, F.; Zhang, Y.; He, J.; Yang, R. Thermal degradation and aging behavior of polytriazole polyethylene oxide-tetrahydrofuran elastomer based on click-chemistry. J. Appl. Polym. Sci. 2020, 48974. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Sbirrazzuoli, N. ICTAC Kinetics Commitee Recommendations for Performing Kinetic Computations on Thermal Analysis Data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Jouyandeh, M.; Paran, S.M.R.; Jannesari, A.; Puglia, D.; Saeb, M.R. Protocol for nonisothermal cure analysis of thermoset composites. Prog. Org. Coat. 2019, 131, 333–339. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Diffusion NMR of molecular cages and capsules. Chem. Soc. Rev. 2015, 44, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Bellachioma, G.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. NMR investigation of non-covalent aggregation of coordination compounds ranging from dimers and ion pairs up to nano-aggregates. Coord. Chem. Rev. 2008, 252, 2224–2238. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Macchioni, A. Extraction of Reliable Molecular Information from Diffusion NMR Spectroscopy: Hydrodynamic Volume or Molecular Mass? Chem. -A Eur. J. 2019, 25, 9930–9937. [Google Scholar] [CrossRef]
- Pages, G.; Gilard, V.; Martino, R.; Malet-Martino, M. Pulsed-field gradient nuclear magnetic resonance measurements (PFG NMR) for diffusion ordered spectroscopy (DOSY) mapping. Analyst 2017, 142, 3771–3796. [Google Scholar] [CrossRef]
- Pregosin, P.S. Applications of NMR diffusion methods with emphasis on ion pairing in inorganic chemistry: A mini-review. Magn. Reson. Chem. 2017, 55, 405–413. [Google Scholar] [CrossRef]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR spectroscopy in supramolecular and combinatorial chemistry: An old parameter—New insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, G.; Li, J.; Luo, Y. Preparation and properties of semi-interpenetrating networks combined by thermoplastic polyurethane and a thermosetting elastomer. New J. Chem. 2018, 42, 3087–3096. [Google Scholar] [CrossRef]
- Zou, Y.; Yang, R.; Zhai, J. Polytriazole polyether elastomers with widely tunable mechanical properties: The role of network structure and crystallization behavior. J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef]
- Lan, Y.; Li, D.; Zhai, J.; Yang, R. Molecular Dynamics Simulation on the Binder of Ethylene Oxide-Tetrahydrofuran Copolyether Cross-Linked with N100. Ind. Eng. Chem. Res. 2015, 54, 3563–3569. [Google Scholar] [CrossRef]
- Le Gresley, A.; Broadberry, G.; Robertson, C.; Peron, J.-M.R.; Robinson, J.; O’Leary, S. Application of pure shift and diffusion NMR for the characterisation of crude and processed pyrolysis oil. J. Anal. Appl. Pyrolysis 2019, 140, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Dudas, E.F.; Bodor, A. Quantitative, Diffusion NMR Based Analytical Tool To Distinguish Folded, Disordered, and Denatured Biomolecules. Anal. Chem. 2019, 91, 4929–4933. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Laryea, E.; Wilhelm, M.; Luy, B.; Nirschl, H.; Guthausen, G. Diffusion in Polymer Solutions: Molecular Weight Distribution by PFG-NMR and Relation to SEC. Macromol. Chem. Phys. 2017, 218. [Google Scholar] [CrossRef]
- Berry, G.C. Translational frictional constant of comb-branched polymers. J. Polym. Sci. Part A-2 Polym. Phys. 1968, 6, 1551. [Google Scholar] [CrossRef]
- Ajroldi, G.; Marchionni, G.; Pezzin, G. The viscosity-molecular weight relationships for diolic perfluoropolyethers. Polymer 1999, 40, 4163–4164. [Google Scholar] [CrossRef]
- Wanger, C.D.; Riggs, W.M.; Davis, L.E.; Moulder, G.F. Handbook of X-ray Photoelectron Spectroscopy; Heyden & Son Ltd.: London, UK, 1979. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physical Polymer Science, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 8, 891–892. [Google Scholar] [CrossRef]
- Fu, X.; Fan, X.; Ju, X.; Qi, X.; Li, J.; Yu, H. Molecular dynamic simulations on the interaction between an HTPE polymer and energetic plasticizers in a solid propellant. RSC Adv. 2015, 5, 52844–52851. [Google Scholar] [CrossRef]
- Tran, T.V.; Couture, G.; Do, L.H. Evaluation of dicopper azacryptand complexes in aqueous CuAAC reactions and their tolerance toward biological thiols. Dalton Trans. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Tang, W.; Li, Y.; He, J.; Guo, X.; Yang, R. The Effect of Glycidyl Azide Polymer Grafted Tetrafunctional Isocyanate on Polytriazole Polyethylene Oxide-Tetrahydrofuran Elastomer and its Propellant Properties. Polymers 2020, 12, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y. Research on the Mechanical Properties of Azide Polyuretgane Elastomers and Its Propellant. Ph.D. Thesis, Nanjing University of Science and Technology, Nanjing, China, 2018. [Google Scholar]
Scheme 1.
Synthesis of polytriazole polyethylene oxide−tetrahydrofuran (PTPET) elastomer.

Figure 1.
Chemical models of Alkynyl-terminated polyethylene oxide−tetrahydrofuran (ATPET) & A3 (bis-(2,2-dinitropropy) acetal (BDNPA)/bis-(2,2-dinitropropyl) formal (BDNPF) = 1/1) (left) and ATPET & Bu-NENA (right).
Figure 1.
Chemical models of Alkynyl-terminated polyethylene oxide−tetrahydrofuran (ATPET) & A3 (bis-(2,2-dinitropropy) acetal (BDNPA)/bis-(2,2-dinitropropyl) formal (BDNPF) = 1/1) (left) and ATPET & Bu-NENA (right).

Figure 2.
Temporal evolution of the 1H-NMR spectra of the PTPET+ A3 (A) and PTPET+ Bu-NENA (B) systems.
Figure 2.
Temporal evolution of the 1H-NMR spectra of the PTPET+ A3 (A) and PTPET+ Bu-NENA (B) systems.

Figure 3.
DOSY-NMR spectral analysis of PTPET/plasticizer mixtures at different times. (A) A3 system, (B) Bu-NENA system, (C) Relationships between diffusion coefficient and time for the above two reaction systems.
Figure 3.
DOSY-NMR spectral analysis of PTPET/plasticizer mixtures at different times. (A) A3 system, (B) Bu-NENA system, (C) Relationships between diffusion coefficient and time for the above two reaction systems.

Figure 4.
Viscosity curves of PTPET with A3 (A) and Bu-NENA (B) after different times.

Figure 5.
Viscosity data and fitting lines for PTPET with Bu-NENA and A3 at various time.

Figure 6.
Gel permeation chromatography (GPC) curves of the uncrosslinked polymer in PTPET after different times, (A) A3 and (B) Bu-NENA.
Figure 6.
Gel permeation chromatography (GPC) curves of the uncrosslinked polymer in PTPET after different times, (A) A3 and (B) Bu-NENA.

Figure 7.
X-ray photoelectron spectroscopy (XPS) analysis of pure catalyst and Bu-NENA soaked catalyst.
Figure 7.
X-ray photoelectron spectroscopy (XPS) analysis of pure catalyst and Bu-NENA soaked catalyst.

Figure 8.
Radial distribution functions (RDFs) for different atoms of PTPET & A3 (A) / PTPET & Bu-NENA (B).
Figure 8.
Radial distribution functions (RDFs) for different atoms of PTPET & A3 (A) / PTPET & Bu-NENA (B).

Figure 9.
TG (A) and derivative thermogravimetric analysis (DTG) (B) curves of Bu-NENA and catalyst.
Figure 9.
TG (A) and derivative thermogravimetric analysis (DTG) (B) curves of Bu-NENA and catalyst.

Figure 10.
Stress-strain curves of PTPET (including Bu-NENA) with GAP (Mn = 4000 g/mol).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Analysis data of the uncrosslinked polymer samples in PTPET.
| A3 Sample | Mn (g/mol) | Mw (g/mol) | PDI | Bu-NENA Sample | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|---|---|---|---|
| 1 d | 5341 | 10,159 | 1.66 | 1 d | 5277 | 9957 | 1.68 |
| 3 d | 5498 | 10,379 | 1.68 | 3 d | 5343 | 10,748 | 1.69 |
| 4 d | 5915 | 11,535 | 1.77 | 4 d | 5376 | 10,793 | 1.72 |
| 6 d | 6236 | 12,128 | 1.95 | 6 d | 5897 | 11,494 | 1.81 |
| 8 d | 6343 | 12,191 | 2.38 | 8 d | 5947 | 11,546 | 1.83 |
Table 2.
Cohesive energy density (CED) and δ of PTPET with Bu-NENA and A3.
| Material | m (Bu-NENA:ATPET) = 1:1 | m (A3/ATPET) = 1:1 | ATPET |
|---|---|---|---|
| CED/(J cm−3) | 426.2 | 423.9 | 338.9 |
| CEDv/(J cm−3) | 335.3 | 297.6 | 296.3 |
| CEDe/(J cm−3) | 74.66 | 110.5 | 263.5 |
| δ/(J cm−3)0.5 | 20.65 | 20.59 | 18.40 |
| δv/(J cm−3)0.5 | 18.31 | 17.25 | 17.21 |
| δe/(J cm−3)0.5 | 8.64 | 10.51 | 5.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hu, J.; Liu, Y.; Cong, K.; He, J.; Yang, R. Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers. Polymers 2020, 12, 1199. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12051199
AMA Style
Hu J, Liu Y, Cong K, He J, Yang R. Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers. Polymers. 2020; 12(5):1199. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12051199
Chicago/Turabian StyleHu, Jinghui, Yina Liu, Kun Cong, Jiyu He, and Rongjie Yang. 2020. "Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers" Polymers 12, no. 5: 1199. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12051199
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.