
Process Design and Optimization towards Digital Twins for HIV-Gag VLP Production in HEK293 Cells, including Purification

 ,
,  , , and
, , and
Abstract
:1. Introduction
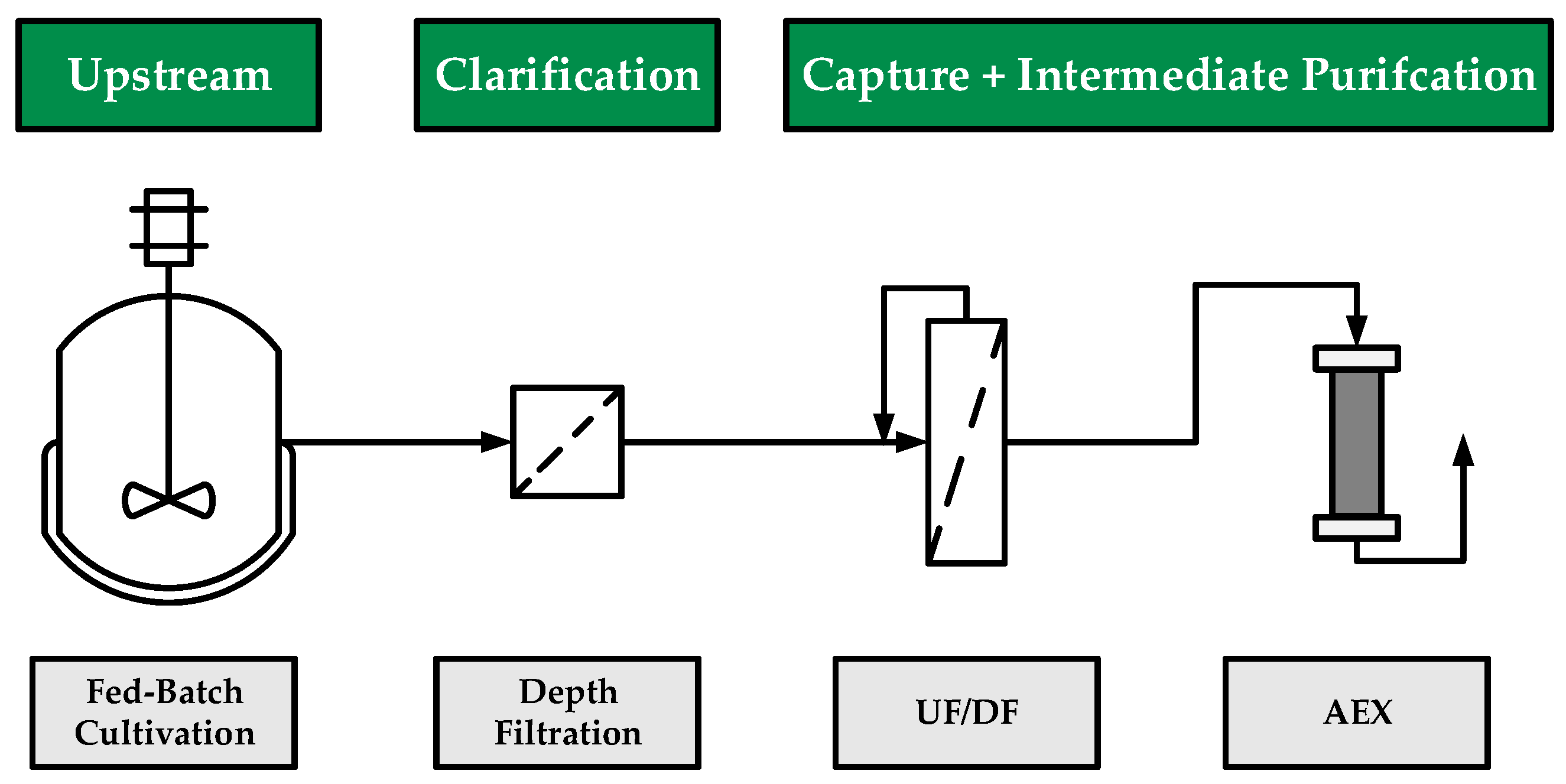
2. Materials and Methods
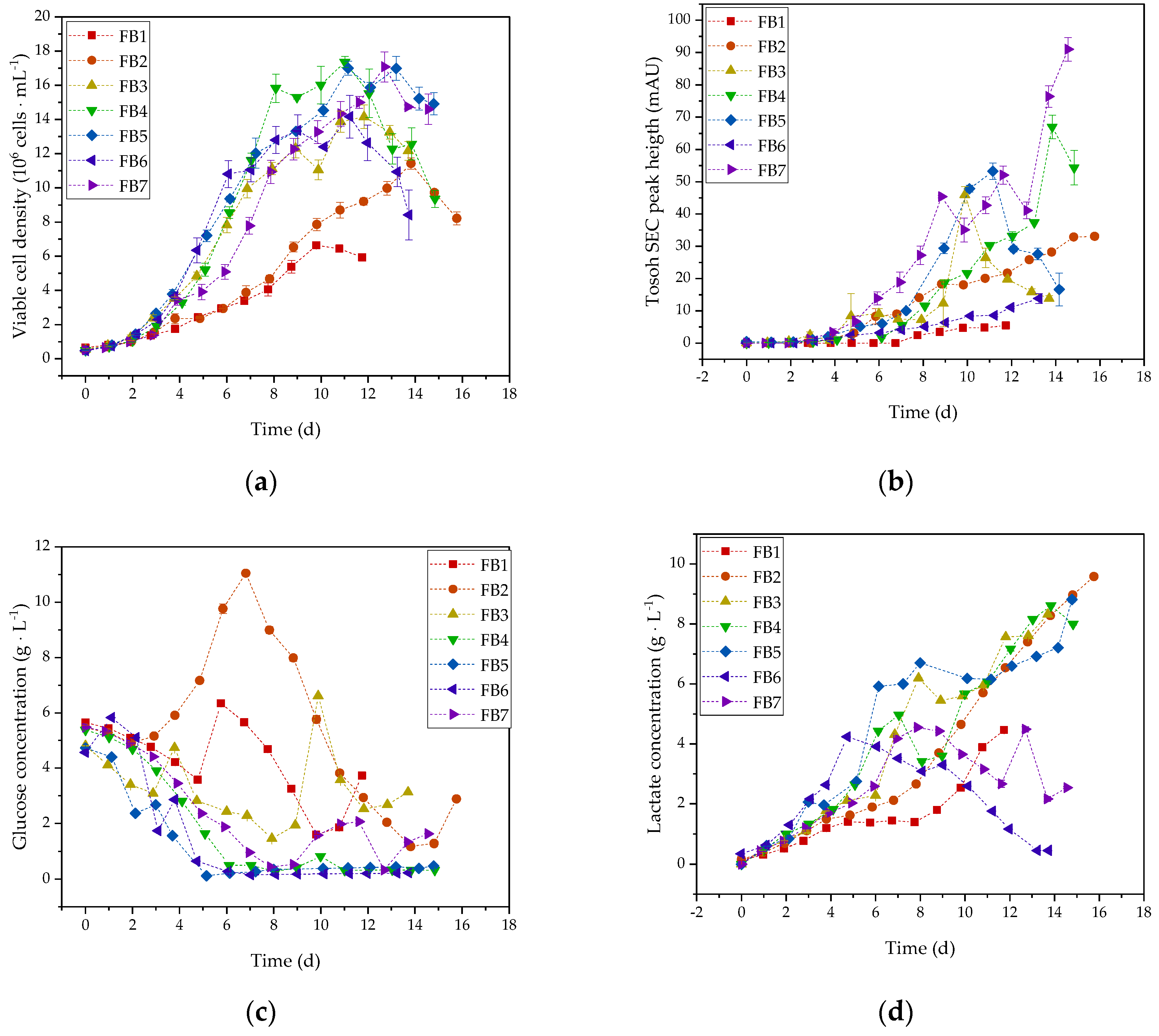
2.1. Fed-Batch Cultivation
2.2. Depth Filtration
2.3. Ultra- and Diafiltration
2.4. Anion-Exchange Chromatography
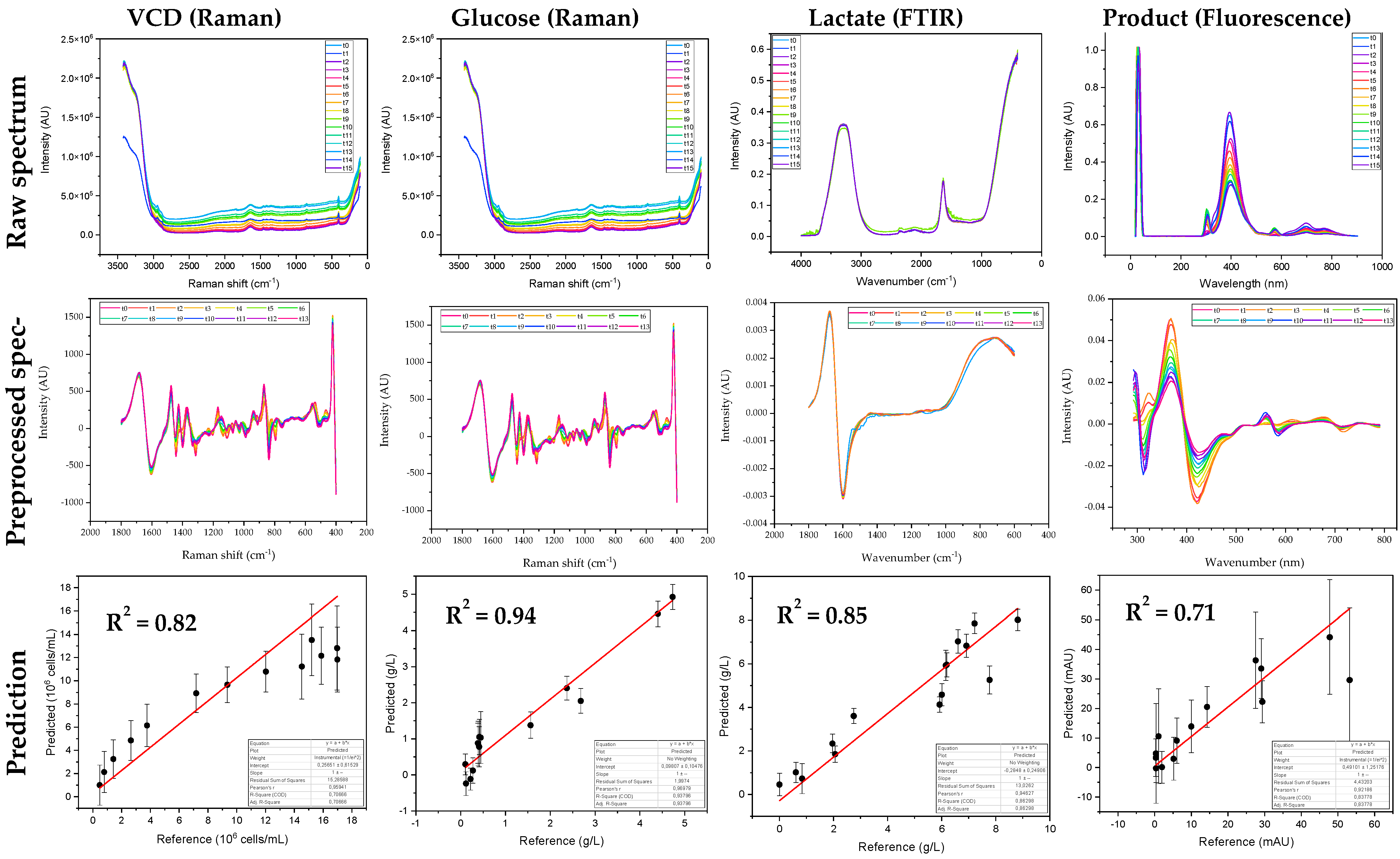
2.5. Processing of Spectral Data
2.6. SEC Analysis
2.7. p24 ELISA
3. Results and Discussion
3.1. Optimization of Cultivation of HIV-Gag Producing 293F Cells
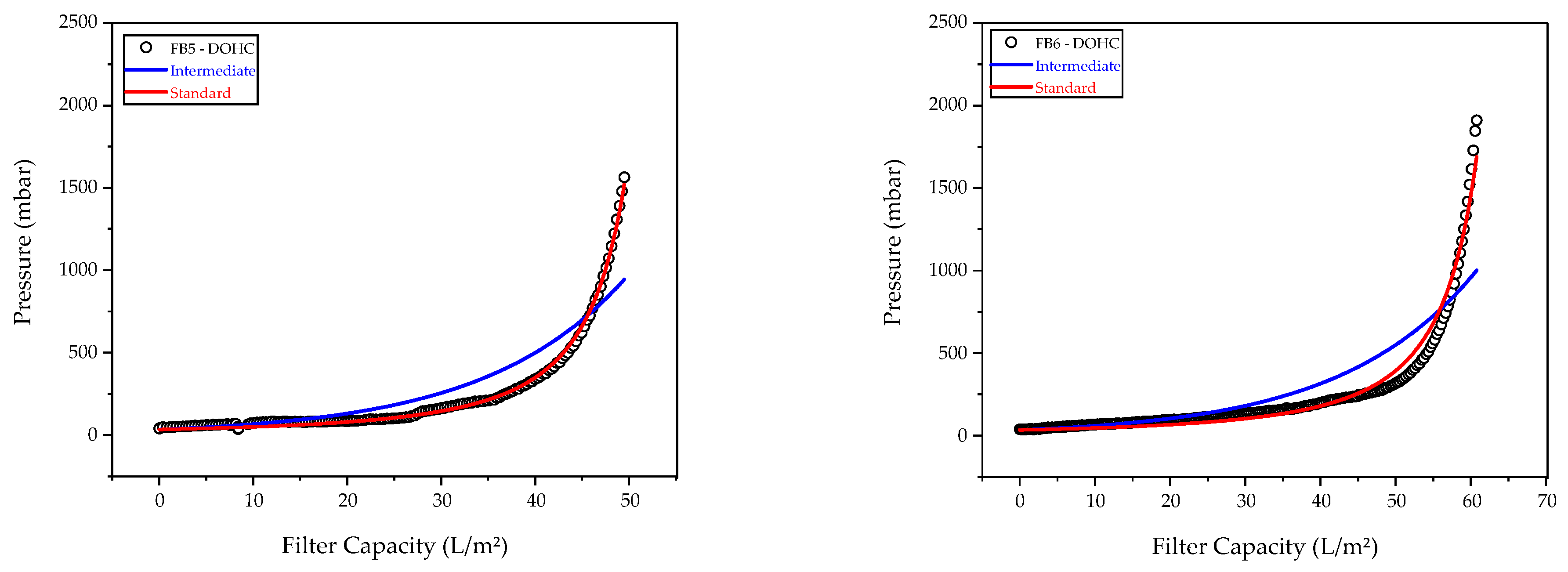
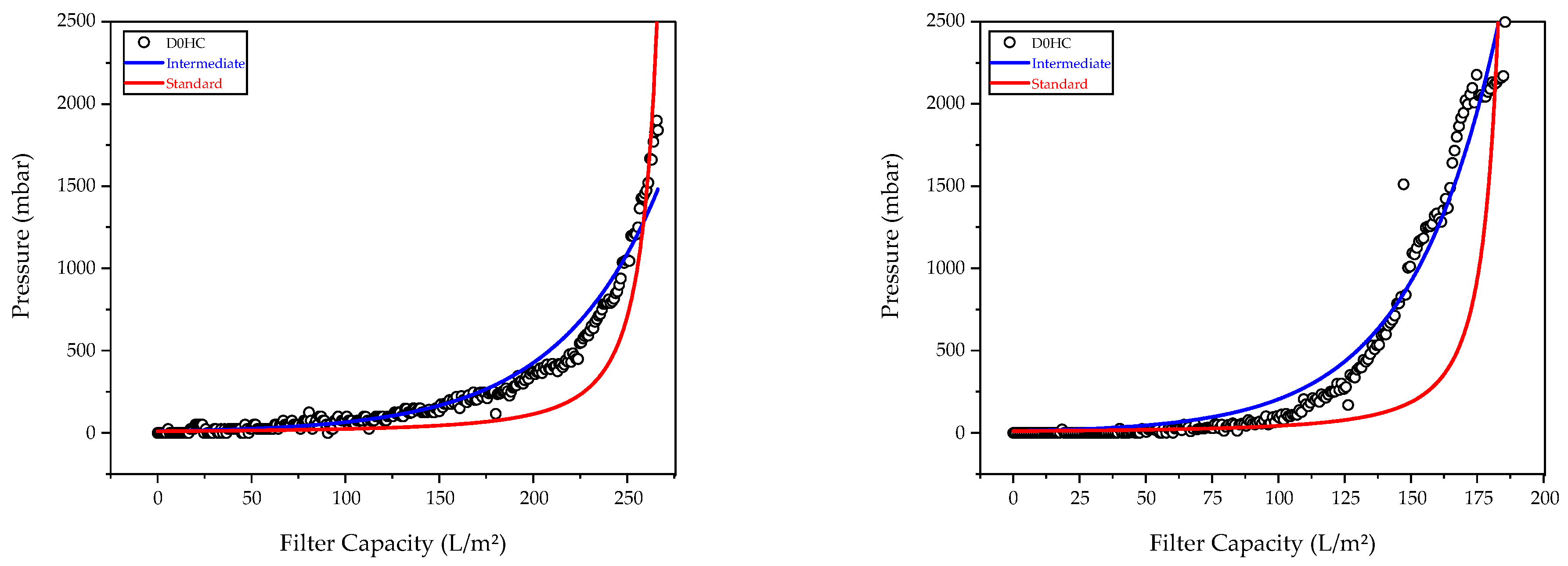
3.2. Characterization of Harvest via Depth Filtration
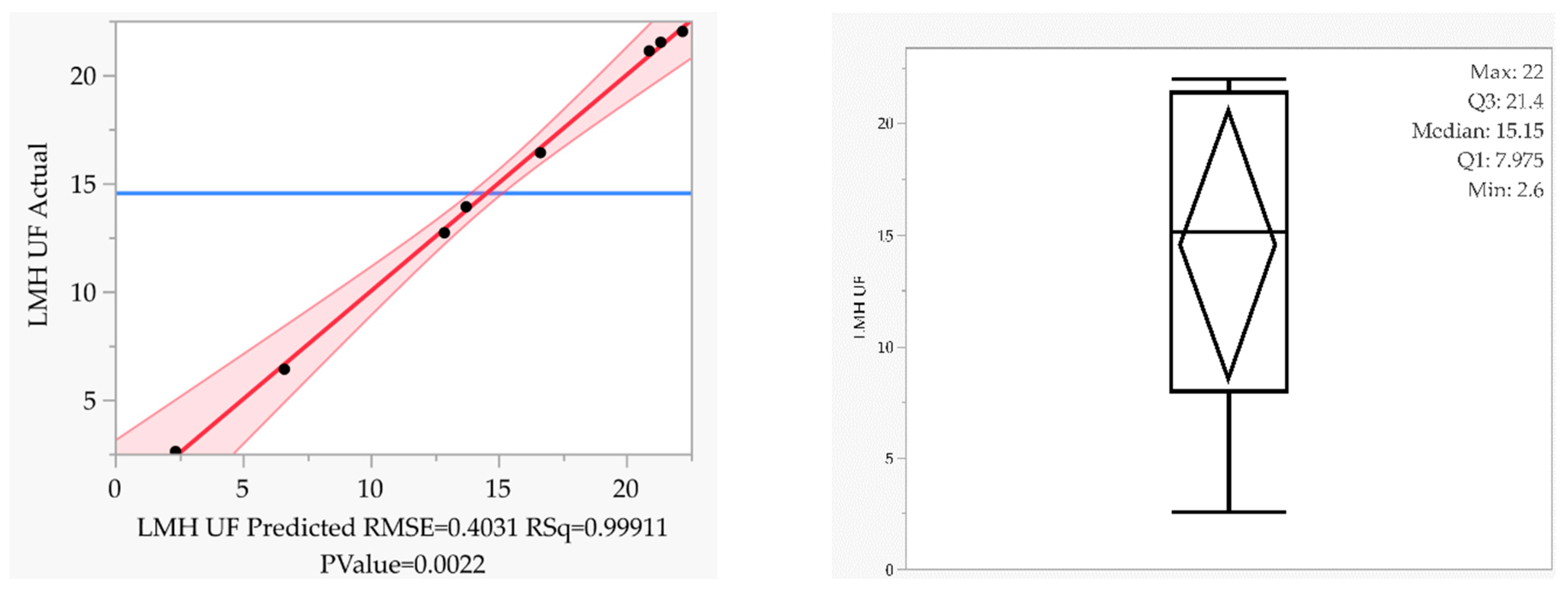
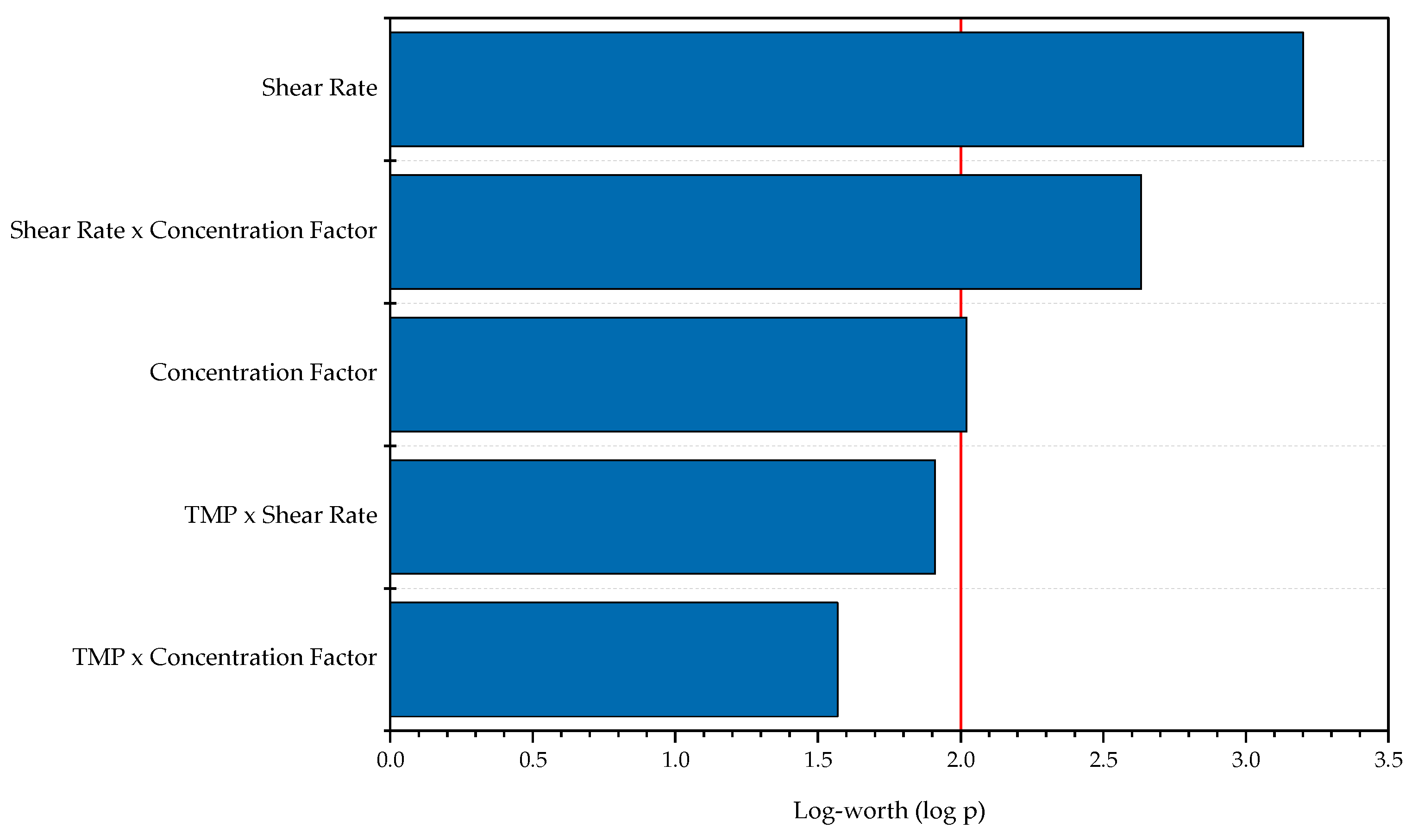
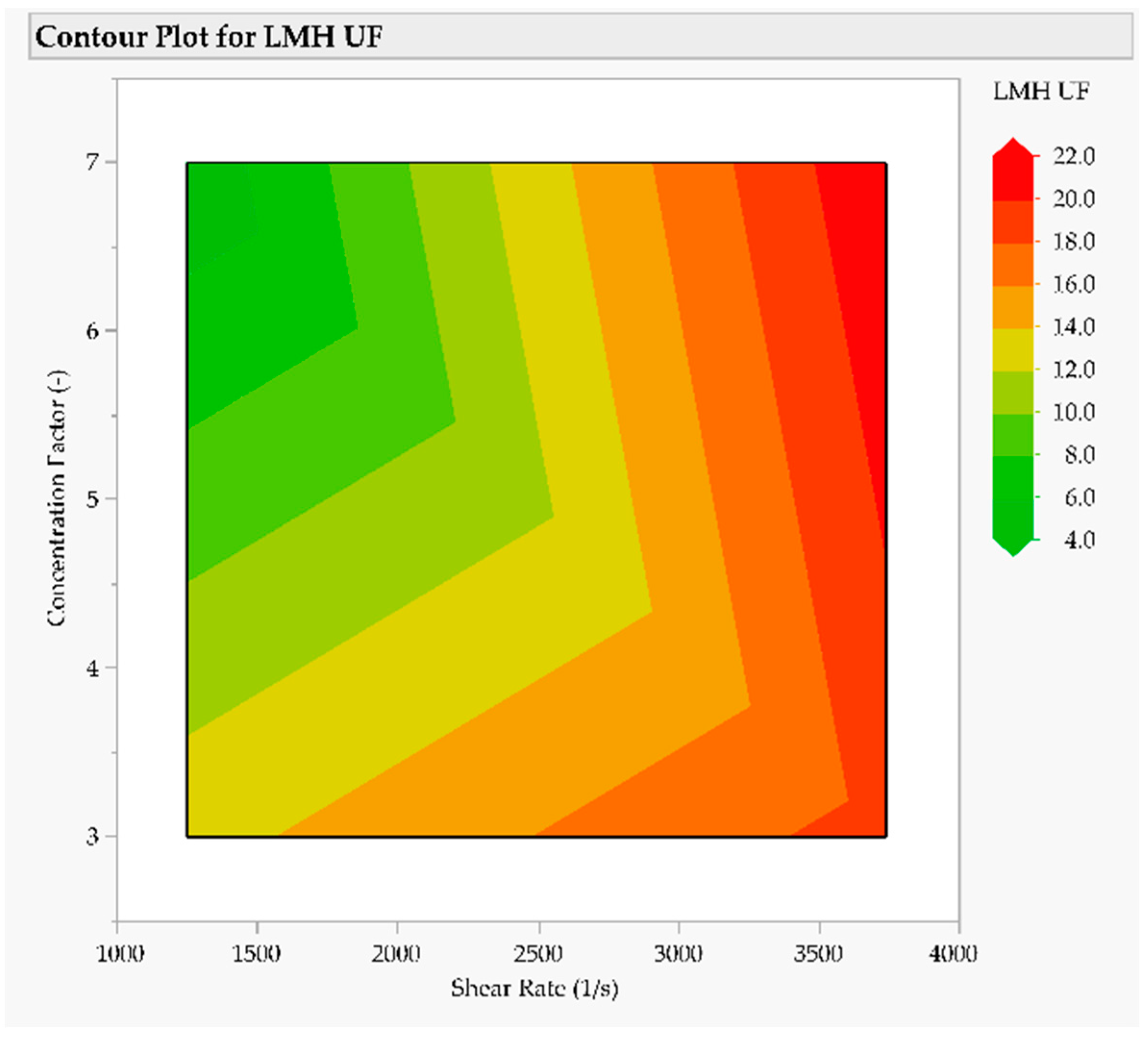
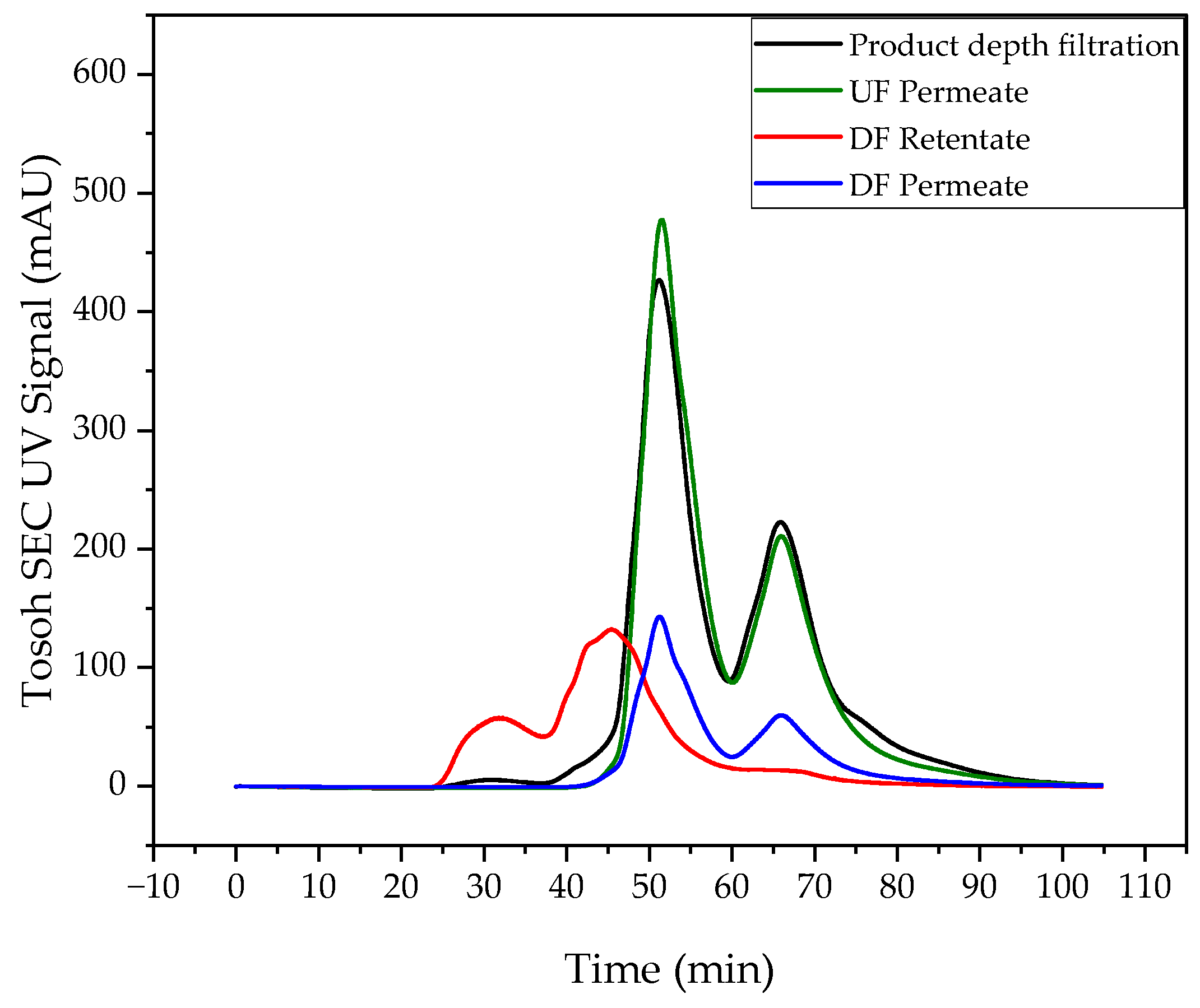
3.3. Intermediate Purification and Concentration via Ultra- and Diafiltration
3.4. Anion-Exchange Chromatography
4. Summary
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flynn, N.M.; Forthal, D.N.; Harro, C.D.; Judson, F.N.; Mayer, K.H.; Para, M.F. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 2005, 191, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Hammonds, J.; Chen, X.; Zhang, X.; Lee, F.; Spearman, P. Advances in methods for the production, purification, and characterization of HIV-1 Gag—Env pseudovirion vaccines. Vaccine 2007, 25, 8036–8048. [Google Scholar] [CrossRef] [PubMed]
- Deml, L.; Speth, C.; Dierich, M.P.; Wolf, H.; Wagner, R. Recombinant HIV-1 Pr55gag virus-like particles: Potent stimulators of innate and acquired immune responses. Mol. Immunol. 2005, 42, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Roldão, A.; Mellado, M.C.M.; Castilho, L.R.; Carrondo, M.J.T.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef]
- Cervera, L.; Gòdia, F.; Tarrés-Freixas, F.; Aguilar-Gurrieri, C.; Carrillo, J.; Blanco, J.; Gutiérrez-Granados, S. Production of HIV-1-based virus-like particles for vaccination: Achievements and limits. Appl. Microbiol. Biotechnol. 2019, 103, 7367–7384. [Google Scholar] [CrossRef]
- Rosengarten, J.F.; Schatz, S.; Wolf, T.; Barbe, S.; Stitz, J. Components of a HIV-1 vaccine mediate virus-like particle (VLP)-formation and display of envelope proteins exposing broadly neutralizing epitopes. Virology 2022, 568, 41–48. [Google Scholar] [CrossRef]
- Ludwig, C.; Wagner, R. Virus-like particles—Universal molecular toolboxes. Curr. Opin. Biotechnol. 2007, 18, 537–545. [Google Scholar] [CrossRef]
- Cruz, P.E.; Cunha, A.; Peixoto, C.C.; Clemente, J.; Moreira, J.L.; Carrondo, M.J. Optimization of the production of virus-like particles in insect cells. Biotechnol. Bioeng. 1998, 60, 408–418. [Google Scholar] [CrossRef]
- Pillay, S.; Meyers, A.; Williamson, A.-L.; Rybicki, E.P. Optimization of chimeric HIV-1 virus-like particle production in a baculovirus-insect cell expression system. Biotechnol. Prog. 2009, 25, 1153–1160. [Google Scholar] [CrossRef]
- Puente-Massaguer, E.; Grau-Garcia, P.; Strobl, F.; Grabherr, R.; Striedner, G.; Lecina, M.; Gòdia, F. Accelerating HIV-1 VLP production using stable High Five insect cell pools. Biotechnol. J. 2021, 16, e2000391. [Google Scholar] [CrossRef]
- Visciano, M.L.; Diomede, L.; Tagliamonte, M.; Tornesello, M.L.; Asti, V.; Bomsel, M.; Buonaguro, F.M.; Lopalco, L.; Buonaguro, L. Generation of HIV-1 Virus-Like Particles expressing different HIV-1 glycoproteins. Vaccine 2011, 29, 4903–4912. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol. Adv. 2012, 30, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Cervera, L.; Gutiérrez-Granados, S.; Martínez, M.; Blanco, J.; Gòdia, F.; Segura, M.M. Generation of HIV-1 Gag VLPs by transient transfection of HEK 293 suspension cell cultures using an optimized animal-derived component free medium. J. Biotechnol. 2013, 166, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Durocher, Y.; Pham, P.L.; St-Laurent, G.; Jacob, D.; Cass, B.; Chahal, P.; Lau, C.J.; Nalbantoglu, J.; Kamen, A. Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J. Virol. Methods 2007, 144, 32–40. [Google Scholar] [CrossRef]
- Kamen, A.; Henry, O. Development and optimization of an adenovirus production process. J. Gene Med. 2004, 6 (Suppl. 1), S184–S192. [Google Scholar] [CrossRef]
- Le Ru, A.; Jacob, D.; Transfiguracion, J.; Ansorge, S.; Henry, O.; Kamen, A.A. Scalable production of influenza virus in HEK-293 cells for efficient vaccine manufacturing. Vaccine 2010, 28, 3661–3671. [Google Scholar] [CrossRef]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef]
- Jalaguier, P.; Turcotte, K.; Danylo, A.; Cantin, R.; Tremblay, M.J. Efficient production of HIV-1 virus-like particles from a mammalian expression vector requires the N-terminal capsid domain. PLoS ONE 2011, 6, e28314. [Google Scholar] [CrossRef]
- Cruz, P.E.; Peixoto, C.C.; Devos, K.; Moreira, J.L.; Saman, E.; Carrondo, M.J. Characterization and downstream processing of HIV-1 core and virus-like-particles produced in serum free medium. Enzym. Microb. Technol. 2000, 26, 61–70. [Google Scholar] [CrossRef]
- Venereo-Sanchez, A.; Simoneau, M.; Lanthier, S.; Chahal, P.; Bourget, L.; Ansorge, S.; Gilbert, R.; Henry, O.; Kamen, A. Process intensification for high yield production of influenza H1N1 Gag virus-like particles using an inducible HEK-293 stable cell line. Vaccine 2017, 35, 4220–4228. [Google Scholar] [CrossRef]
- Negrete, A.; Pai, A.; Shiloach, J. Use of hollow fiber tangential flow filtration for the recovery and concentration of HIV virus-like particles produced in insect cells. J. Virol. Methods 2014, 195, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, M.J.; Soares, R.; Azevedo, A.M.; Chu, V.; Tover, A.; Conde, J.P.; Aires-Barros, M.R. Optimization and miniaturization of aqueous two phase systems for the purification of recombinant human immunodeficiency virus-like particles from a CHO cell supernatant. Sep. Purif. Technol. 2015, 154, 27–35. [Google Scholar] [CrossRef]
- Turpeinen, D.G.; Joshi, P.U.; Kriz, S.A.; Kaur, S.; Nold, N.M.; O’Hagan, D.; Nikam, S.; Masoud, H.; Heldt, C.L. Continuous purification of an enveloped and non-enveloped viral particle using an aqueous two-phase system. Sep. Purif. Technol. 2021, 269, 118753. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Berger, E.; Aguilar, P.P.; Schneider, T.A.; Kramberger, P.; Tover, A.; Nöbauer, K.; Razzazi-Fazeli, E.; et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J. Chromatogr. A 2016, 1455, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Segura, M.M.; Mangion, M.; Gaillet, B.; Garnier, A. New developments in lentiviral vector design, production and purification. Expert Opin. Biol. Ther. 2013, 13, 987–1011. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, I.; Lorenzo, E.; Bernier, A.; Cervera, L.; Gòdia, F.; Kamen, A. A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles. Vaccines 2021, 9, 1154. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, O.; Sampat, C.; Bhalode, P.; Ramachandran, R.; Ierapetritou, M. Digital Twins in Pharmaceutical and Biopharmaceutical Manufacturing: A Literature Review. Processes 2020, 8, 1088. [Google Scholar] [CrossRef]
- Matsunami, K.; Ryckaert, A.; Peeters, M.; Badr, S.; Sugiyama, H.; Nopens, I.; de Beer, T. Analysis of the Effects of Process Parameters on Start-Up Operation in Continuous Wet Granulation. Processes 2021, 9, 1502. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Sixt, M.; Uhlenbrock, L.; Strube, J. Toward a Distinct and Quantitative Validation Method for Predictive Process Modelling—On the Example of Solid-Liquid Extraction Processes of Complex Plant Extracts. Processes 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Uhlenbrock, L.; Sixt, M.; Strube, J. Quality-by-Design (QbD) process evaluation for phytopharmaceuticals on the example of 10-deacetylbaccatin III from yew. Resour. -Effic. Technol. 2017, 3, 137–143. [Google Scholar] [CrossRef]
- Schmidt, A.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modeling with Examples of Liquid-Liquid Extraction Processes of Complex Feed Mixtures. Processes 2019, 7, 298. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable Without a Digital-Twin? Processes 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Kornecki, M.; Schmidt, A.; Strube, J. PAT as key-enabling technology for QbD in pharmaceutical manufacturing A conceptual review on upstream and downstream processing. Chim. Oggi-Chem. Today 2018, 36, 44–48. [Google Scholar]
- Helgers, H.; Schmidt, A.; Lohmann, L.J.; Vetter, F.L.; Juckers, A.; Jensch, C.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Towards Autonomous Operation by Advanced Process Control—Process Analytical Technology for Continuous Biologics Antibody Manufacturing. Processes 2021, 9, 172. [Google Scholar] [CrossRef]
- Buijs, J.; Norde, W.; Lichtenbelt, J.W.T. Changes in the Secondary Structure of Adsorbed IgG and F(ab′) 2 Studied by FTIR Spectroscopy. Langmuir 1996, 12, 1605–1613. [Google Scholar] [CrossRef]
- Wasalathanthri, D.P.; Feroz, H.; Puri, N.; Hung, J.; Lane, G.; Holstein, M.; Chemmalil, L.; Both, D.; Ghose, S.; Ding, J.; et al. Real-time monitoring of quality attributes by in-line Fourier transform infrared spectroscopic sensors at ultrafiltration and diafiltration of bioprocess. Biotechnol. Bioeng. 2020, 117, 3766–3774. [Google Scholar] [CrossRef]
- Berry, B.; Moretto, J.; Matthews, T.; Smelko, J.; Wiltberger, K. Cross-scale predictive modeling of CHO cell culture growth and metabolites using Raman spectroscopy and multivariate analysis. Biotechnol. Prog. 2015, 31, 566–577. [Google Scholar] [CrossRef]
- Biechele, P.; Busse, C.; Solle, D.; Scheper, T.; Reardon, K. Sensor systems for bioprocess monitoring. Eng. Life Sci. 2015, 15, 469–488. [Google Scholar] [CrossRef]
- ICH Expert Working Group. Riskmanagement (Q9): ICH Harmonised Tripartite Guideline; ICH Expert Working Group: Geneva, Switzerland, 2005. [Google Scholar]
- ICH Expert Working Group. Pharmaceutical Quality System (Q10): ICH Harmonised Tripartite Guideline; ICH Expert Working Group: Geneva, Switzerland, 2008. [Google Scholar]
- ICH Expert Working Group. Pharmaceuticel Development Q8(R2): ICH Harmonised Tripartite Guideline; ICH Expert Working Group: Geneva, Switzerland, 2009. [Google Scholar]
- Qin, S.J.; Badgwell, T.A. A survey of industrial model predictive control technology. Control. Eng. Pract. 2003, 11, 733–764. [Google Scholar] [CrossRef]
- Morari, M.; Lee, J.H. Model predictive control: Past, present and future. Comput. Chem. Eng. 1999, 23, 667–682. [Google Scholar] [CrossRef]
- Camacho, E.F.; Alba, C.B. Model Predictive Control; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 0857293982. [Google Scholar]
- Garcia, C.E.; Prett, D.M.; Morari, M. Model predictive control: Theory and practice—A survey. Automatica 1989, 25, 335–348. [Google Scholar] [CrossRef]
- Dittmar, R. Advanced Process Control: PID-Basisregelungen, Vermaschte Regelungsstrukturen, Softsensoren, Model Predictive Control; DE GRUYTER: Berlin, Germany; Boston, MA, USA, 2017; ISBN 9783110499582. [Google Scholar]
- Keerthi, S.S.; Gilbert, E.G. Optimal infinite-horizon feedback laws for a general class of constrained discrete-time systems: Stability and moving-horizon approximations. J. Optim. Theory Appl. 1988, 57, 265–293. [Google Scholar] [CrossRef] [Green Version]
- Song, I.-H.; Amanullah, M.; Erdem, G.; Mazzotti, M.; Rhee, H.-K. Experimental implementation of identification-based optimizing control of a simulated moving bed process. J. Chromatogr. A 2006, 1113, 60–73. [Google Scholar] [CrossRef]
- Sommeregger, W.; Sissolak, B.; Kandra, K.; von Stosch, M.; Mayer, M.; Striedner, G. Quality by control: Towards model predictive control of mammalian cell culture bioprocesses. Biotechnol. J. 2017, 12, 1600546. [Google Scholar] [CrossRef] [Green Version]
- Fanali, S. Editorial on “Simulated moving bed chromatography for the separation of enantiomers” by A. Rajendran, G. Paredes and M. Mazzotti. J. Chromatogr. A 2009, 1216, 708. [Google Scholar] [CrossRef]
- Diederich, P.; Hansen, S.K.; Oelmeier, S.A.; Stolzenberger, B.; Hubbuch, J. A sub-two minutes method for monoclonal antibody-aggregate quantification using parallel interlaced size exclusion high performance liquid chromatography. J. Chromatogr. A 2011, 1218, 9010–9018. [Google Scholar] [CrossRef]
- Rüdt, M.; Andris, S.; Schiemer, R.; Hubbuch, J. Factorization of preparative protein chromatograms with hard-constraint multivariate curve resolution and second-derivative pretreatment. J. Chromatogr. A 2019, 1585, 152–160. [Google Scholar] [CrossRef]
- Abu-Absi, N.R.; Kenty, B.M.; Cuellar, M.E.; Borys, M.C.; Sakhamuri, S.; Strachan, D.J.; Hausladen, M.C.; Li, Z.J. Real time monitoring of multiple parameters in mammalian cell culture bioreactors using an in-line Raman spectroscopy probe. Biotechnol. Bioeng. 2011, 108, 1215–1221. [Google Scholar] [CrossRef]
- Claßen, J.; Aupert, F.; Reardon, K.F.; Solle, D.; Scheper, T. Spectroscopic sensors for in-line bioprocess monitoring in research and pharmaceutical industrial application. Anal. Bioanal. Chem. 2017, 409, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Kornecki, M.; Strube, J. Process Analytical Technology for Advanced Process Control in Biologics Manufacturing with the Aid of Macroscopic Kinetic Modeling. Bioengineering 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.M.; Kessler, J.-M.; Salou, P.; Menezes, J.C.; Peinado, A. Monitoring mAb cultivations with in-situ raman spectroscopy: The influence of spectral selectivity on calibration models and industrial use as reliable PAT tool. Biotechnol. Prog. 2018, 34, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Kornecki, M.; Strube, J. Accelerating Biologics Manufacturing by Upstream Process Modelling. Processes 2019, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Rolinger, L.; Rüdt, M.; Diehm, J.; Chow-Hubbertz, J.; Heitmann, M.; Schleper, S.; Hubbuch, J. Multi-attribute PAT for UF/DF of Proteins—Monitoring Concentration, particle sizes, and Buffer Exchange. Anal. Bioanal. Chem. 2020, 412, 2123–2136. [Google Scholar] [CrossRef]
- Jungbauer, A.; Dürauer, A.; Walch, N.; SAUER, D.; Scharl-Hirsch, T.; Melcher, M.; Leisch, F. Real-Time Monitoring of Product Purification. International Patent Application No. WO2017174580A1, 4 April 2017. [Google Scholar]
- Klepzig, L.S.; Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Digital Twin for Lyophilization by Process Modeling in Manufacturing of Biologics. Processes 2020, 8, 1325. [Google Scholar] [CrossRef]
- Langedijk, J.P.M.; Callendret, B.C.S.; Van Manen, D.; Krarup, A.; Stitz, J.; Wegmann, F.; Vellinga, J. Human Immunodeficiency Virus Antigens, Vectors, Compositions, and Methods of Use Thereof. International Patent Application No. AU2019203468B2, 21 May 2020. [Google Scholar]
- Langedijk, J.P.M.; Van Manen, D.; Vellinga, J.; Wegmann, F.; Callendret, B.C.S.; Krarup, A.; Stitz, J. Recombinant Adenoviruses Encoding Mosaic Human Immunodeficiency Virus (HIV) Env, Gag, and Pol antigens. U.S. Patent Application No. 10,973,907, 13 April 2021. [Google Scholar]
- Pereira Aguilar, P.; González-Domínguez, I.; Schneider, T.A.; Gòdia, F.; Cervera, L.; Jungbauer, A. At-line multi-angle light scattering detector for faster process development in enveloped virus-like particle purification. J. Sep. Sci. 2019, 42, 2640–2649. [Google Scholar] [CrossRef]
- Esbensen, K.H. Multivariate Data Analysis—In Practice: An Introduction to Multivariate Data Analysis and Experimental Design, 5th ed.; CAMO Analytics: Bedford, MA, USA, 2012; ISBN 8299333032. [Google Scholar]
- MilliporeSigma. Clarification of Mammalian Cell Cultures by Depth Filtration; MilliporeSigma: Burlington, MA, USA, 2017. [Google Scholar]
- Iritani, E.; Katagiri, N. Developments of Blocking Filtration Model in Membrane Filtration. KONA Powder Part. J. 2016, 33, 179–202. [Google Scholar] [CrossRef] [Green Version]
- Repligen. Product Information—Midikros: Shear Conditions Graph. 2021. Available online: https://store.repligen.com/collections/midikros-hollow-fiber-filters/products/midikros-20cm-0-05um-ps-0-5mm-fll-x-fll-1-pk (accessed on 7 December 2021).
- Compound Interest. Analytical Chemistry—Infrared (IR) Spectroscopy. 2015. Available online: https://www.compoundchem.com/2015/02/05/irspectroscopy/ (accessed on 8 December 2021).

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cultivation | Cell Line | Base Medium | Feed Medium | Glucose Target Concentration after Feed Addition (g/L) |
|---|---|---|---|---|
| 1 | 293FwtGag | PM | PM | 6 |
| 2 | 293FwtGag | PM | Feed Supplement | 6 |
| 3 | 293FwtGag | SMD | Feed Supplement | 6 |
| 4 | 293FMos1.Gag | SMD | Feed Supplement | 3–3.5 |
| 5 | 293FMos1.Gag | SMD | Feed Supplement | 2.5–3 |
| 6 | 293FMos1.Gag | SMD | Cell Boost 6 | 2.5–3 |
| 7 | 293FMos1.Gag/Mos2S.Env | SMD | Feed Supplement | 2.5–3 |
| Filter | Cut-Off (µm) | Manufacturer |
|---|---|---|
| Millistak+D0HC | 0.55–9 | Merck KGaA, Darmstadt, Germany |
| PDP8 | 6–30 | Pall Corporation, Port Washington, NY, USA |
| PDK5 | 1.5–20 | |
| PDH4 | 0.5–15 | |
| Bio20 | 0.4–1 |
| Experiment | TMP (bar) | Shear Rate (s−1) | Concentration Factor (-) |
|---|---|---|---|
| 1 | 1.5 | 3738 | 7 |
| 2 | 1.5 | 3738 | 3 |
| 3 | 1.5 | 1249 | 3 |
| 4 | 0.5 | 1249 | 3 |
| 5 | 0.5 | 3738 | 7 |
| 6 | 0.5 | 1249 | 7 |
| 7 | 0.5 | 3738 | 3 |
| 8 | 1.5 | 1249 | 7 |
| Model | Equation | Blocking Constant (dm−1) | Adj. R2 | Feed |
|---|---|---|---|---|
| Intermediate | 0.06713 ± 2.3 × 10−4 | 0.852 | FB5 | |
| 0.05564 ± 1.6 × 10−4 | 0.761 | FB6 | ||
| Standard | 0.03436 ± 2.79 × 10−4 | 0.997 | FB5 | |
| 0.02824 ± 5.63 × 10−4 | 0.975 | FB6 |
| Model | Equation | Blocking Constant (dm−1) | Adj R2 | Feed |
|---|---|---|---|---|
| Intermediate | 0.01876 ± 8.1 × 10−6 | 0.948 | PC1 | |
| 0.03015 ± 9.4 × 10−6 | 0.976 | PC2 | ||
| Standard | 0.00705 ± 1.28 × 10−6 | 0.792 | PC1 | |
| 0.001025 ± 3.39 × 10−6 | 0.489 | PC2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helgers, H.; Hengelbrock, A.; Schmidt, A.; Rosengarten, J.; Stitz, J.; Strube, J. Process Design and Optimization towards Digital Twins for HIV-Gag VLP Production in HEK293 Cells, including Purification. Processes 2022, 10, 419. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10020419
Helgers H, Hengelbrock A, Schmidt A, Rosengarten J, Stitz J, Strube J. Process Design and Optimization towards Digital Twins for HIV-Gag VLP Production in HEK293 Cells, including Purification. Processes. 2022; 10(2):419. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10020419
Chicago/Turabian StyleHelgers, Heribert, Alina Hengelbrock, Axel Schmidt, Jamila Rosengarten, Jörn Stitz, and Jochen Strube. 2022. "Process Design and Optimization towards Digital Twins for HIV-Gag VLP Production in HEK293 Cells, including Purification" Processes 10, no. 2: 419. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10020419