
Relevance of Particle Size Distribution to Kinetic Analysis: The Case of Thermal Dehydroxylation of Kaolinite


Abstract
:1. Introduction
2. Theoretical Approach
3. Experimental Section
4. Results and Discussion
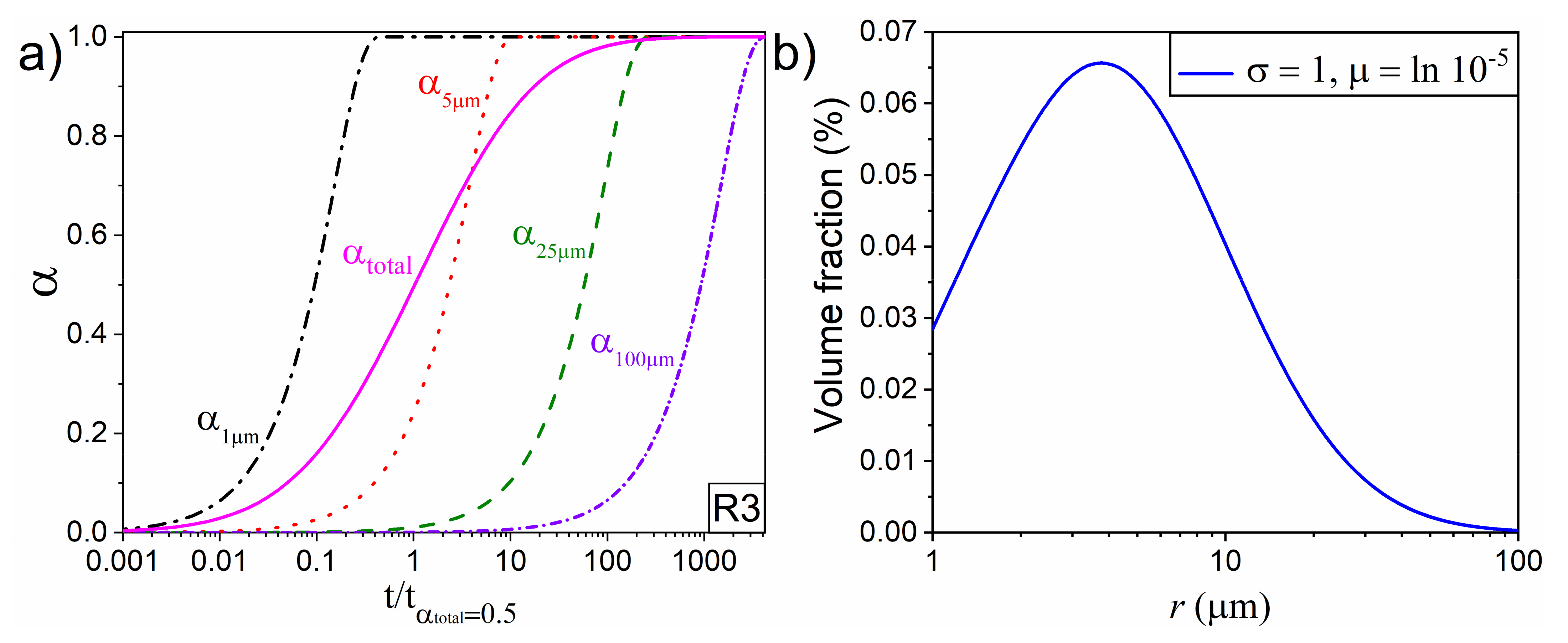
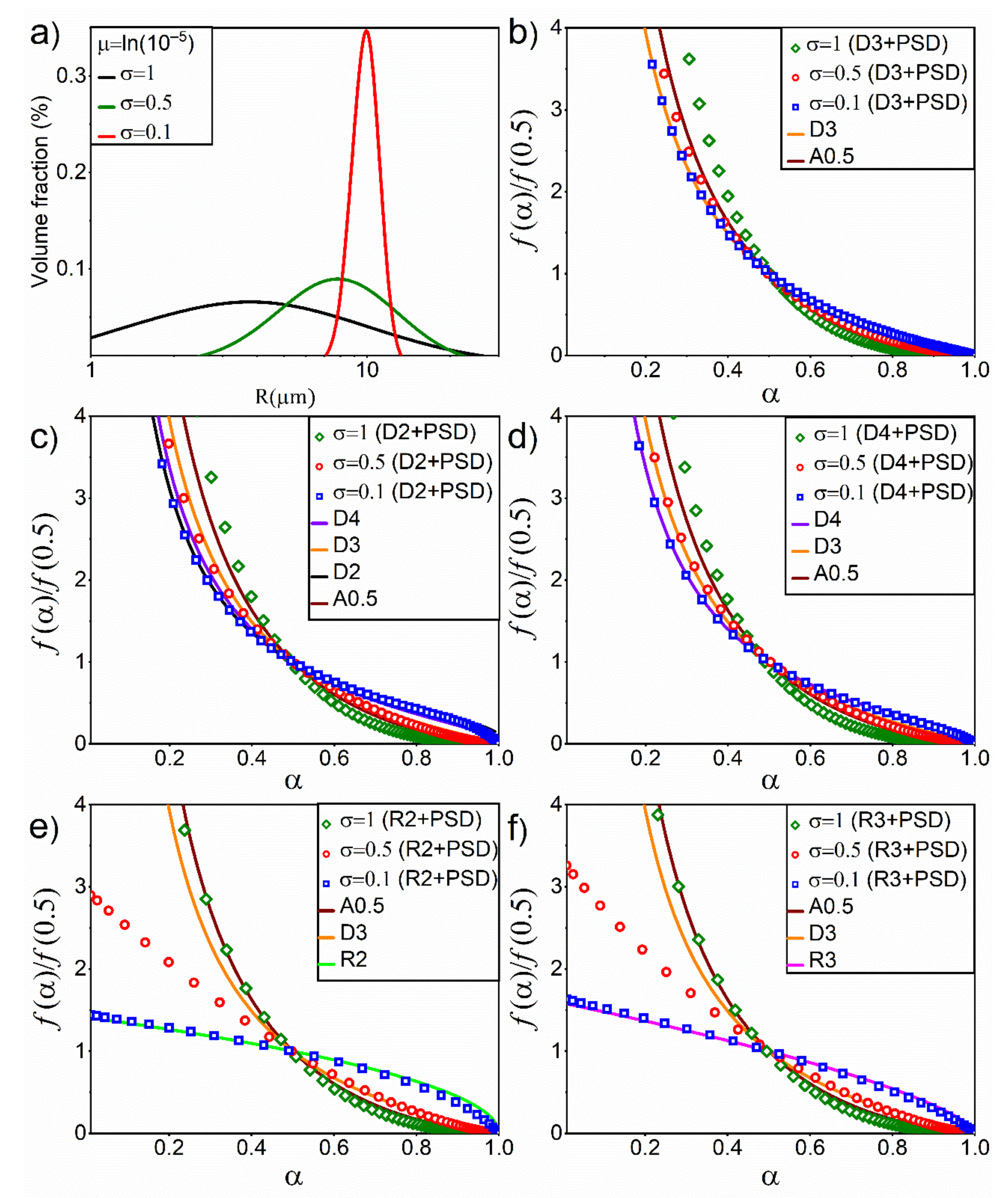
4.1. Effect of PSD in Simulated Linear Heating Experiments
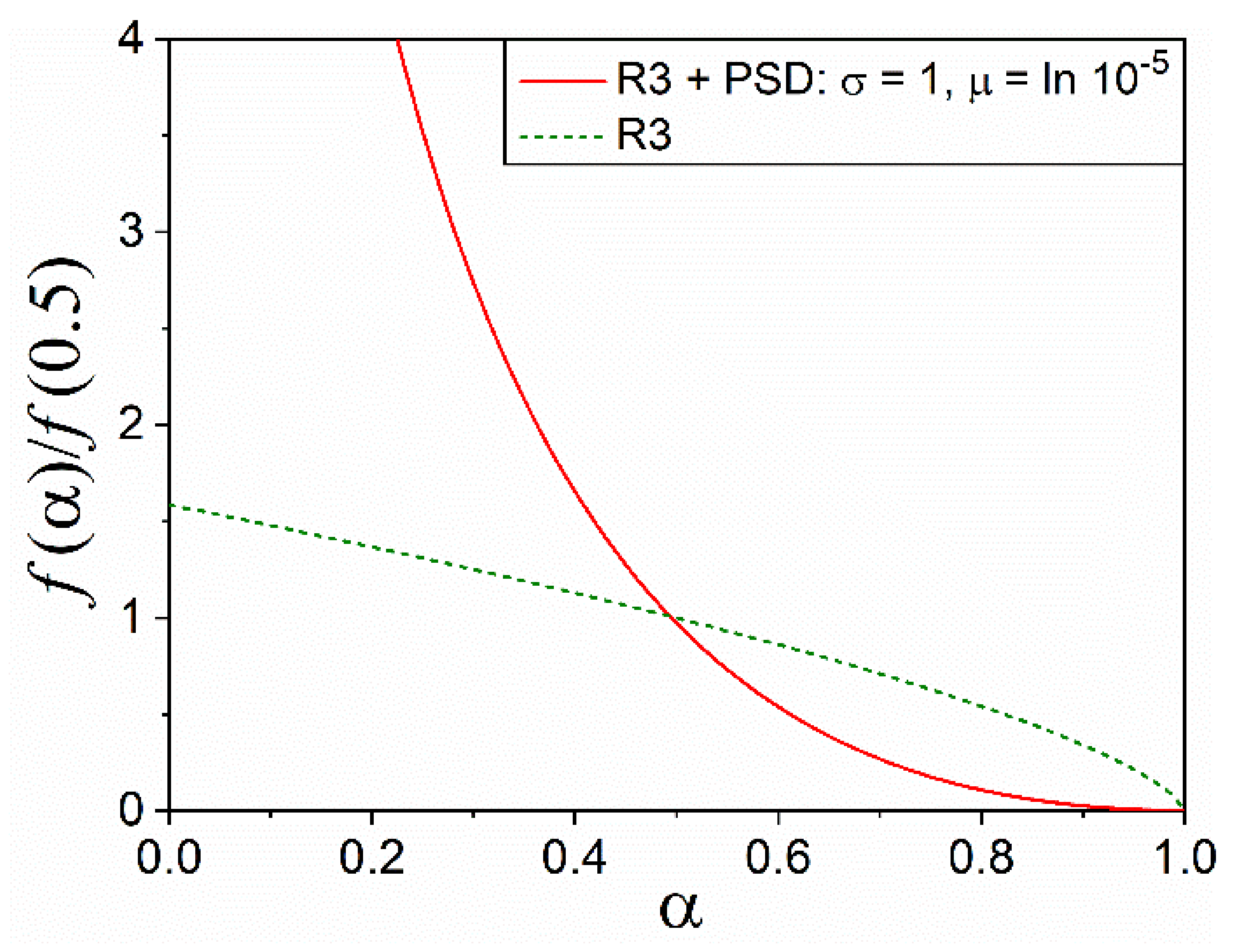
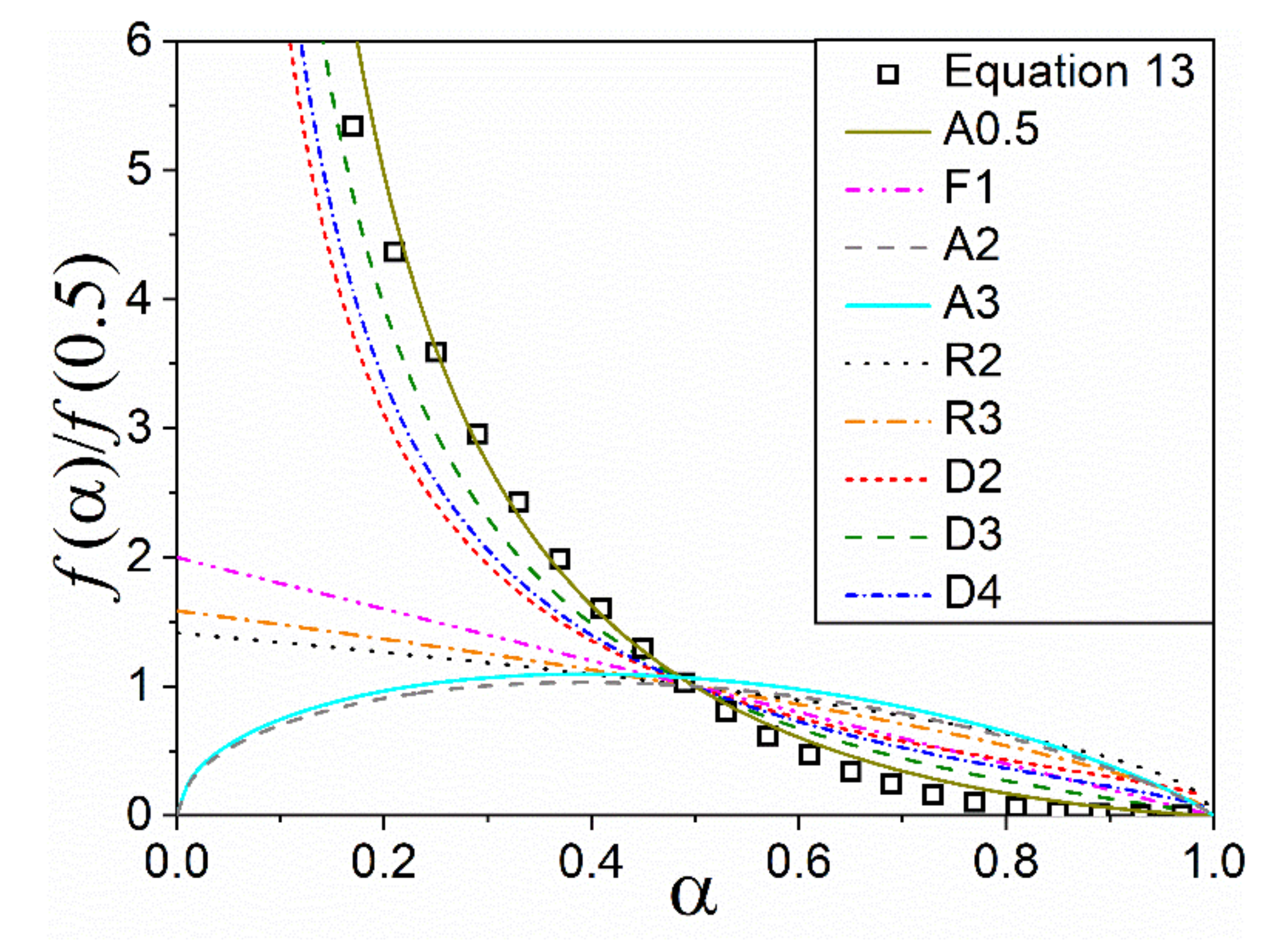
4.2. Effect of PSD on Diffusion and Interface Reaction Models
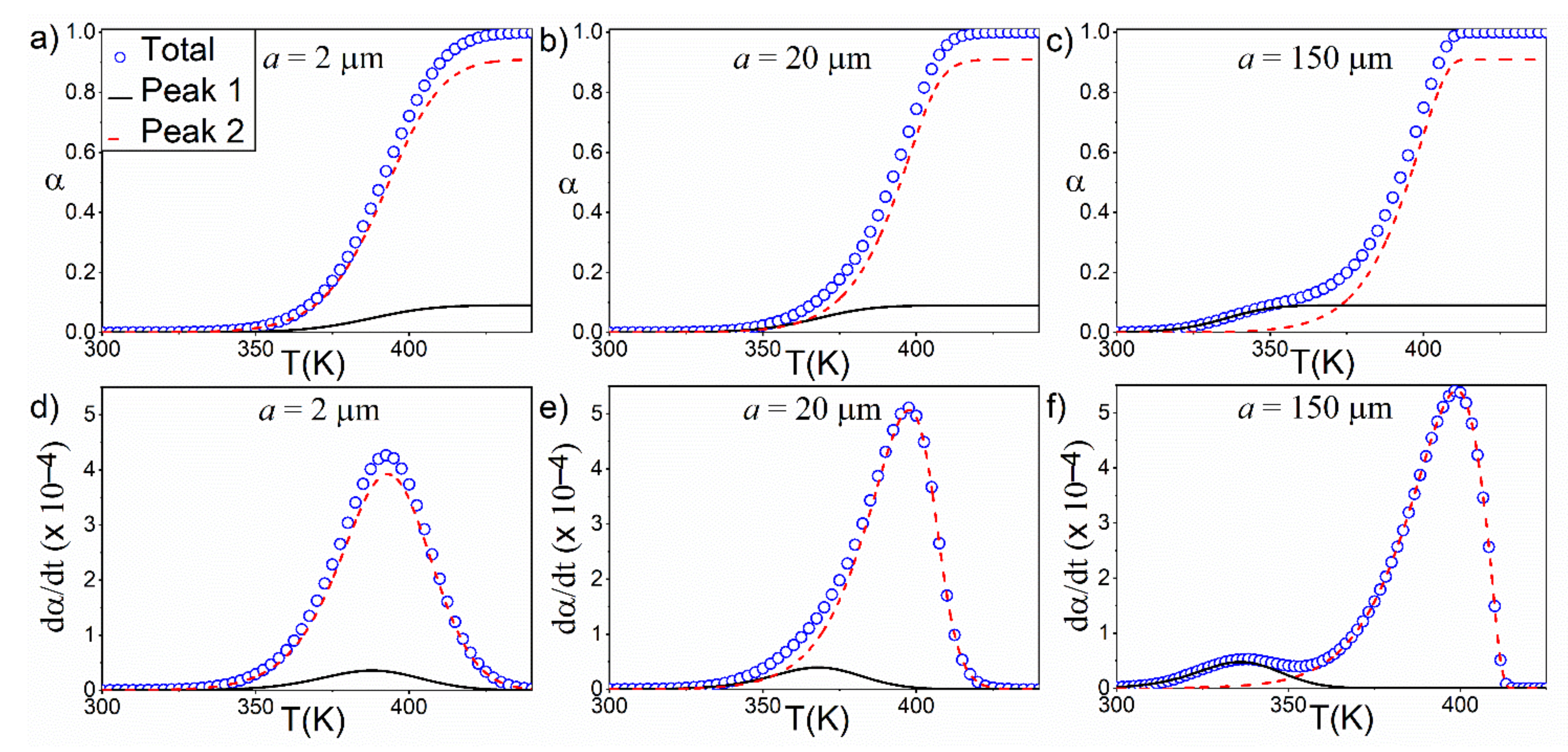
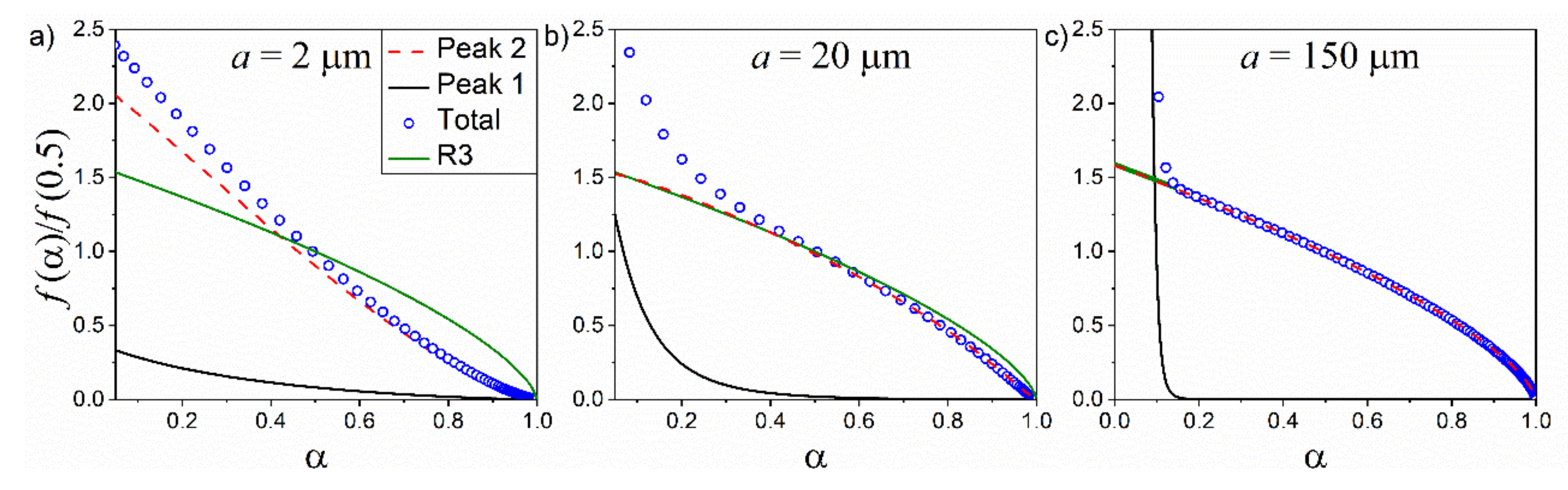
4.3. Effect of a Bimodal PSD in Kinetic Curves
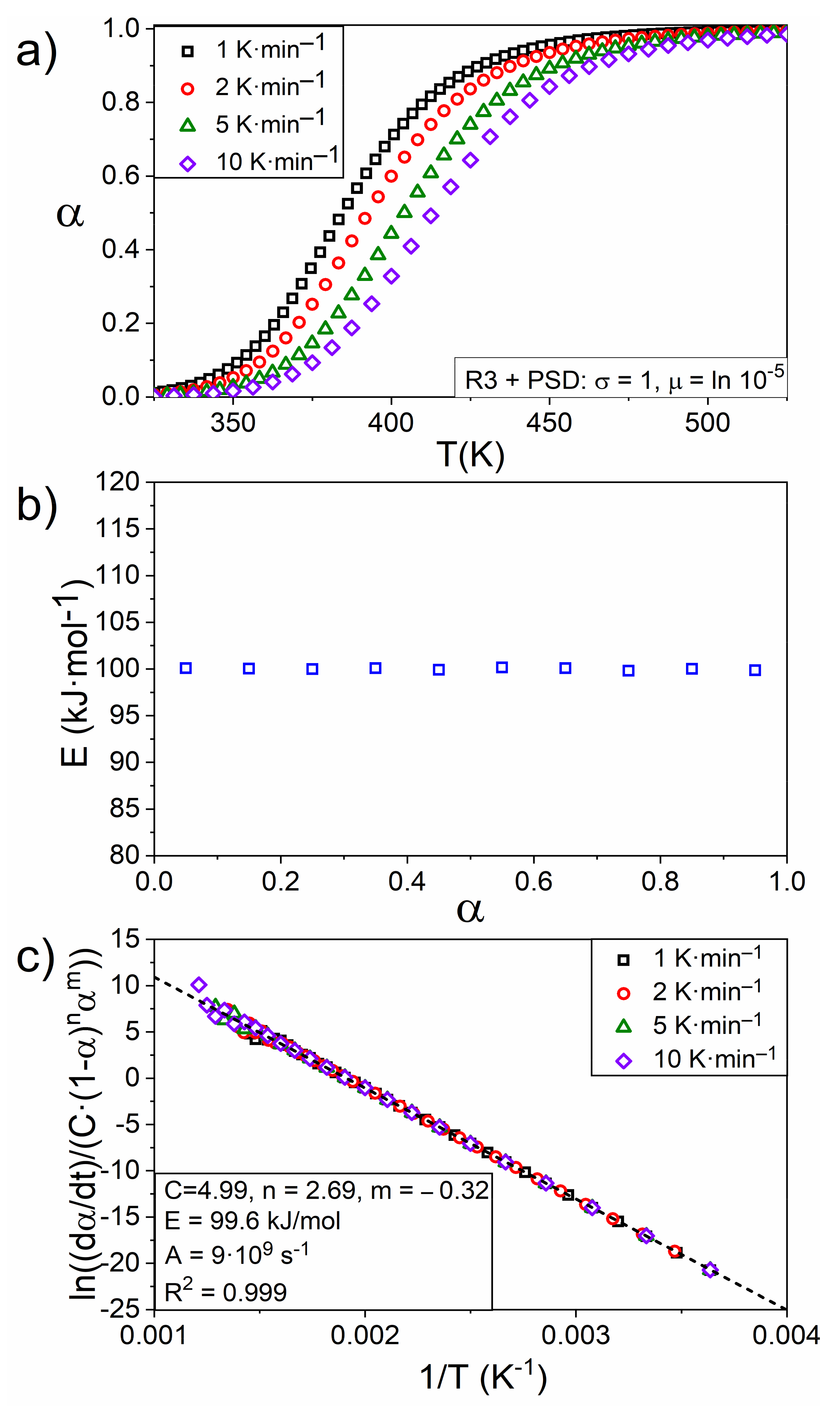
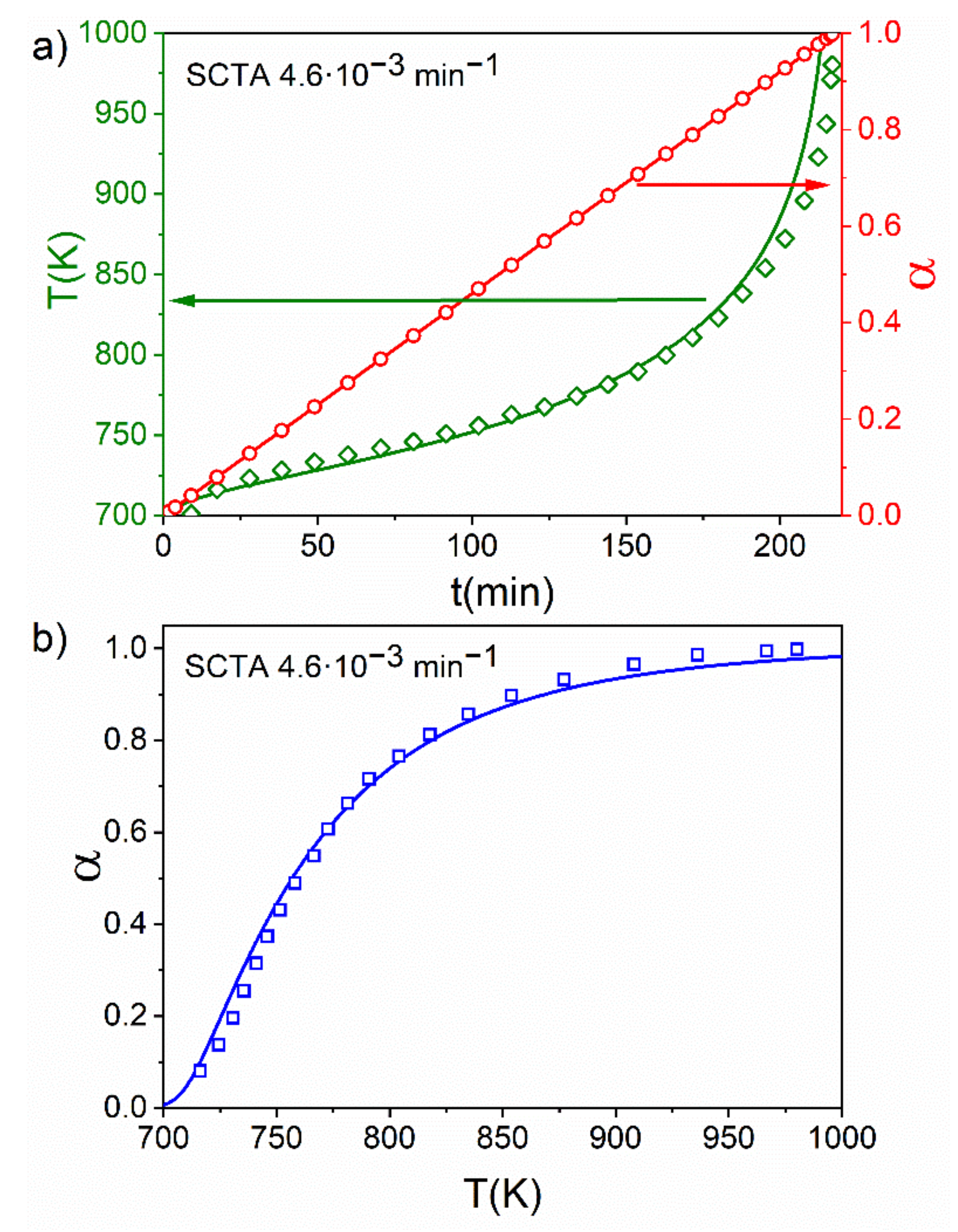
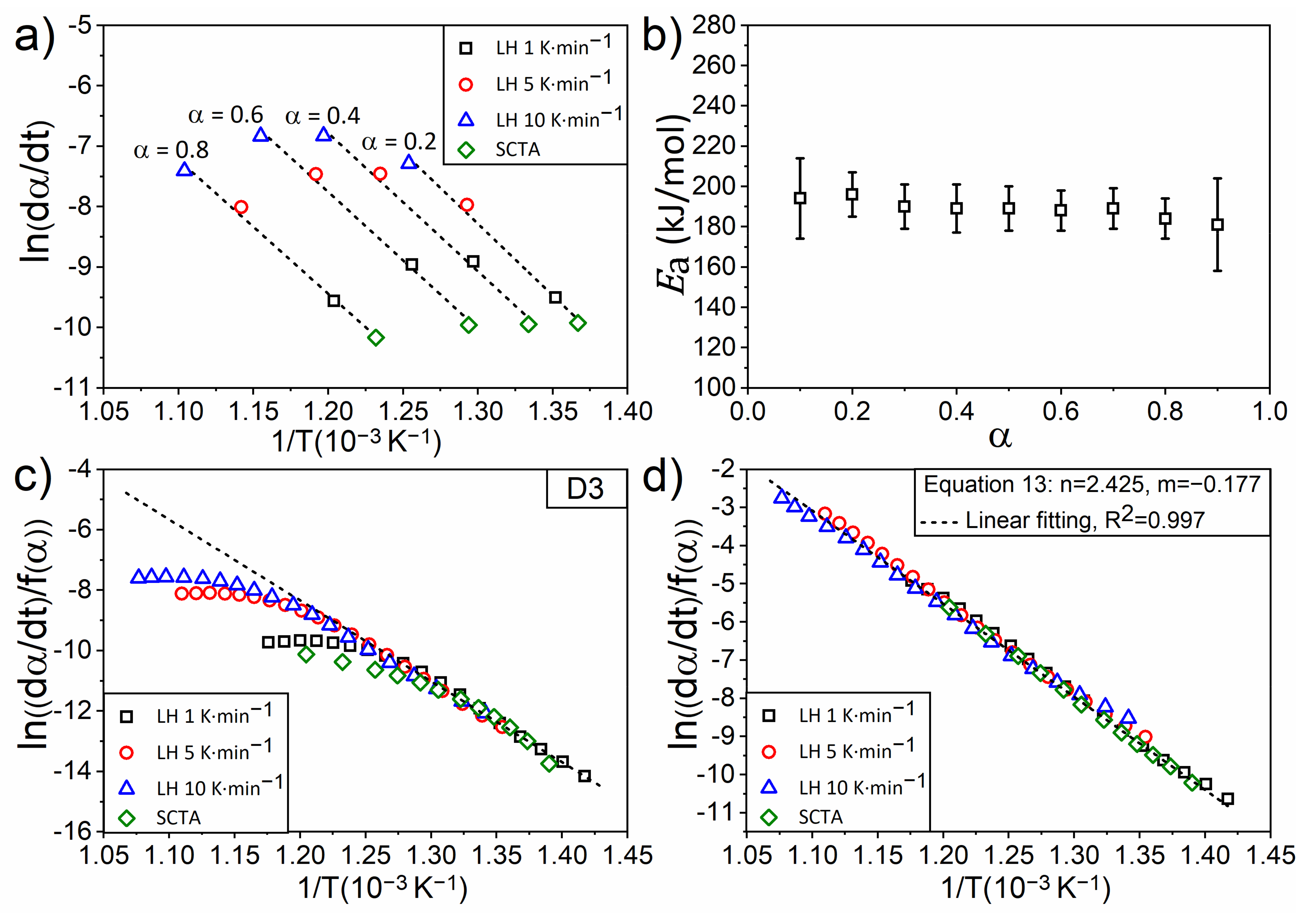
4.4. An Experimental Case: Kinetic of Thermal Dehydroxylation of Kaolinite
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khawam, A.; Flanagan, D.R. Solid-State Kinetic Models: Basics and Mathematical Fundamentals. J. Phys. Chem. B 2006, 110, 17315–17328. [Google Scholar] [CrossRef]
- Galwey, M.E.; Brown, A.K. Background to Thermal Analysis and Calorimetry, In Handbook of Thermal Analysis and Calorimetry: Applications to Inorganic and Miscellaneous Materials; Elservier Science: Amsterdam, The Netherlands, 1998; Volume 1, p. 147. [Google Scholar]
- Koga, N.; Tanaka, H. A physico-geometric approach to the kinetics of solid-state reactions as exemplified by the thermal dehydration and decomposition of inorganic solids. Thermochim. Acta 2002, 388, 41–61. [Google Scholar] [CrossRef]
- Skrdla, P.J. Use of Coupled Rate Equations To Describe Nucleation-and-Branching Rate-Limited Solid-State Processes. J. Phys. Chem. A 2004, 108, 6709–6712. [Google Scholar] [CrossRef]
- Fedunik-Hofman, L.; Bayon, A.; Donne, S.W. Kinetics of Solid-Gas Reactions and Their Application to Carbonate Looping Systems. Energies 2019, 12, 2981. [Google Scholar] [CrossRef] [Green Version]
- Frade, J.R.; Cable, M. Reexamination of the Basic Theoretical Model for the Kinetics of Solid-State Reactions. J. Am. Ceram. Soc. 1992, 75, 1949–1957. [Google Scholar] [CrossRef]
- Cooper, E.A.; Mason, T.O. Mechanism of La2CuO4 Solid-State Powder Reaction by Quantitative XRD and Impedance Spectroscopy. J. Am. Ceram. Soc. 1995, 78, 857–864. [Google Scholar] [CrossRef]
- Sasaki, H. Introduction of Particle-Size Distribution into Kinetics of Solid-State Reaction. J. Am. Ceram. Soc. 1964, 47, 512–516. [Google Scholar] [CrossRef]
- Kapur, P.C. Kinetics of Solid-State Reactions of Particulate Ensembles with Size Distributions. J. Am. Ceram. Soc. 1973, 56, 79–81. [Google Scholar] [CrossRef]
- Miyagi, S. Criticism on Jander’s Equation of Reaction-Rate, Considering Statistical Distribution of Particle Size of Reacting Substance. J. Ceram. Assoc. Jpn. 1951, 59, 132–135. [Google Scholar] [CrossRef] [Green Version]
- Koga, N.; Criado, J.M. Kinetic Analyses of Solid-State Reactions with a Particle-Size Distribution. J. Am. Ceram. Soc. 1998, 81, 2901–2909. [Google Scholar] [CrossRef]
- Urrutia, G.A.; Blesa, M.A. The influence of particle size distribution on the conversion/time profiles under contracting-geometry kinetic regimes. React. Solids 1988, 6, 281–284. [Google Scholar] [CrossRef]
- HMurray, H.H. Traditional and new applications for kaolin, smectite, and palygorskite: A general overview. Appl. Clay Sci. 2000, 17, 207–221. [Google Scholar] [CrossRef]
- Massaro, M.; Colletti, C.G.; Lazzara, G.; Riela, S. The Use of Some Clay Minerals as Natural Resources for Drug Carrier Applications. J. Funct. Biomater. 2018, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- ERuiz-Hitzky, E.; Aranda, P.; Darder, M. Hybrid and Biohybrid Materials Based on Layered Clays. In Tailored Organic-Inorganic Materials, 1st ed.; Brunet, E., Colón, J.L., Clearfield, A., Eds.; Instituto de Ciencia de Materiales de Madrid, ICMM-CSIC Cantoblanco: Madrid, Spain, 2015; pp. 245–297. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, L.; Cheng, H. Rational design of kaolinite-based photocatalytic materials for environment decontamination. Appl. Clay Sci. 2021, 208, 106098. [Google Scholar] [CrossRef]
- Franco, F.; Pérez-Maqueda, L.; Pérez-Rodríguez, J. The effect of ultrasound on the particle size and structural disorder of a well-ordered kaolinite. J. Colloid Interface Sci. 2004, 274, 107–117. [Google Scholar] [CrossRef]
- Belviso, C.; Cavalcante, F.; Lettino, A.; Fiore, S. A and X-type zeolites synthesised from kaolinite at low temperature. Appl. Clay Sci. 2013, 80–81, 162–168. [Google Scholar] [CrossRef]
- Rashad, A.M. Metakaolin as cementitious material: History, scours, production and composition—A comprehensive overview. Constr. Build. Mater. 2013, 41, 303–318. [Google Scholar] [CrossRef]
- Nawaz, M.; Heitor, A.; Sivakumar, M. Geopolymers in construction—Recent developments. Constr. Build. Mater. 2020, 260, 120472. [Google Scholar] [CrossRef]
- Criado, J.M.; Ortega, A.; Real, C.; De Torres, E.T. Re-examination of the kinetics of the thermal dehydroxylation of kaolinite. Clay Miner. 1984, 19, 653–661. [Google Scholar] [CrossRef]
- Bellotto, M.; Gualtieri, A.; Artioli, G.; Clark, S.M. Kinetic study of the kaolinite-mullite reaction sequence. Part I: Kaolinite dehydroxylation. Phys. Chem. Miner. 1995, 22, 207–217. [Google Scholar] [CrossRef]
- Redfern, S.A.T. The kinetics of dehydroxylation of kaolinite. Clay Miner. 1987, 22, 447–456. [Google Scholar] [CrossRef]
- Gasparini, E.; Tarantino, S.; Ghigna, P.; Riccardi, M.P.; Cedillo-González, E.I.; Siligardi, C.; Zema, M. Thermal dehydroxylation of kaolinite under isothermal conditions. Appl. Clay Sci. 2013, 80–81, 417–425. [Google Scholar] [CrossRef]
- Ortega, A.; Gotor, F.J.; Macías, M. The Multistep Nature of the Kaolinite Dehydroxylation: Kinetics and Mechanism. J. Am. Ceram. Soc. 2010, 93, 197–203. [Google Scholar] [CrossRef]
- Murray, P.; White, J. Kinetics of the Thermal Dehydration of Clays. Trans. Br. Ceram. Soc. 1955, 54, 137–150. [Google Scholar]
- Frost, R.L.; Vassallo, A.M. The Dehydroxylation of the Kaolinite Clay Minerals using Infrared Emission Spectroscopy. Clays Clay Miner. 1996, 44, 635–651. [Google Scholar] [CrossRef]
- Ortega, A.; Rouquérol, F.; Akhouayri, S.; Laureiro, Y.; Rouquerol, J. Kinetical study of the thermolysis of kaolinite between −30° and 1000 °C by controlled rate evolved gas analysis. Appl. Clay Sci. 1993, 8, 207–214. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Hu, Y. Investigation of the thermal behaviour and decomposition kinetics of kaolinite. Clay Miner. 2015, 50, 199–209. [Google Scholar] [CrossRef]
- Ptáček, P.; Frajkorová, F.; Soukal, F.; Opravil, T. Kinetics and mechanism of three stages of thermal transformation of kaolinite to metakaolinite. Powder Technol. 2014, 264, 439–445. [Google Scholar] [CrossRef]
- Ptáček, P.; Soukal, F.; Opravil, T.; Havlica, J.; Brandštetr, J. The kinetic analysis of the thermal decomposition of kaolinite by DTG technique. Powder Technol. 2011, 208, 20–25. [Google Scholar] [CrossRef]
- KNahdi, K.; Llewellyn, P.; Rouquérol, F.; Ariguib, N.; Ayedi, M. Controlled Rate Thermal Analysis of kaolinite dehydroxylation: Effect of water vapour pressure on the mechanism. Thermochim. Acta 2002, 390, 123–132. [Google Scholar] [CrossRef]
- Allison, E.B. The dtermination of specific heats of reaction of clay minerals by thermal analysis. Silic. Ind. 1954, 19, 363–373. [Google Scholar]
- Cabrera, J.; Eddleston, M. Kinetics of dehydroxylation and evaluation of the crystallinity of kaolinite. Thermochim. Acta 1983, 70, 237–247. [Google Scholar] [CrossRef]
- Ondro, T.; Húlan, T.; Vitázek, I. Non-Isothermal Kinetic Analysis of the Dehydroxylation of Kaolinite in Dynamic Air Atmosphere. Acta Technol. Agric. 2017, 20, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Saikia, D.N.; Sengupta, P.; Gogoi, P.; Borthakur, P.C. Kinetics of dehydroxylation of kaolin in presence of oil field effluent treatment plant sludge. Appl. Clay Sci. 2002, 22, 93–102. [Google Scholar] [CrossRef]
- Levy, J.H.; Hurst, H. Kinetics of dehydroxylation, in nitrogen and water vapour, of kaolinite and smectite from Australian Tertiary oil shale. Fuel 1993, 72, 873–877. [Google Scholar] [CrossRef]
- Ptáček, P.; Kubátová, D.; Havlica, J.; Brandštetr, J.; Soukal, F.; Opravil, T. The non-isothermal kinetic analysis of the thermal decomposition of kaolinite by thermogravimetric analysis. Powder Technol. 2010, 204, 222–227. [Google Scholar] [CrossRef]
- Brindley, G.W.; Sharp, J.H.; Patterson, J.H.; Narahari, B.N. Kinetics and Mechanism of Dehydroxylation Processes, I. Temperature and Vapor Pressure Dependence of Dehydroxylation of Kaolinite. Am. Mineral. 1967, 52, 201–211. [Google Scholar]
- Redaoui, D.; Sahnoune, F.; Heraiz, M.; Belhouchet, H.; Fatmi, M. Thermal decomposition kinetics of Algerian Tamazarte kaolinite by thermogravimetric analysis. Trans. Nonferrous Met. Soc. China 2017, 27, 1849–1855. [Google Scholar] [CrossRef]
- Horváth, I. Kinetics and compensation effect in kaolinite dehydroxylation. Thermochim. Acta 1985, 85, 193–198. [Google Scholar] [CrossRef]
- Dion, P.; Alcover, J.-F.; Bergaya, F.; Ortega, A.; Llewellyn, P.L.; Rouquérol, F. Kinetic study by controlled-transformation rate thermal analysis of the dehydroxylation of kaolinite. Clay Miner. 1998, 33, 269–276. [Google Scholar] [CrossRef]
- Zhang, X.L.Z.; Zhong, Y.; Wang, L.; Liao, G.; Wang, R.; Li, Z.; Li, J. Kinetic analysis of non-isothermal dehydroxylation of kaolinite based on isoconversional and multivariate non-linear regression methods. Kuangwu Yanshi 2019, 39, 1–7. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.; Sanchez, P.; Criado, J. Evaluation of the integral methods for the kinetic study of thermally stimulated processes in polymer science. Polymer 2005, 46, 2950–2954. [Google Scholar] [CrossRef] [Green Version]
- Perez-Rodriguez, J.L.; Duran, A.; Jiménez, P.E.S.; Franquelo, M.L.; Perejón, A.; Pascual-Cosp, J.; Pérez-Maqueda, L.A. Study of the Dehydroxylation-Rehydroxylation of Pyrophyllite. J. Am. Ceram. Soc. 2010, 93, 2392–2398. [Google Scholar] [CrossRef] [Green Version]
- Rouquerol, J. Thermal analysis: Sample-controlled techniques. In Encyclopedia of Analytical Science; Elsevier: Amsterdam, The Netherlands, 2019; pp. 17–321. [Google Scholar]
- Pérez-Maqueda, L.A.; Ortega, A.; Criado, J.M. The use of master plots for discriminating the kinetic model of solid state reactions from a single constant-rate thermal analysis (CRTA) experiment. Thermochim. Acta 1996, 277, 165–173. [Google Scholar] [CrossRef]
- Chopra, G.S.; Real, C.; Alcalá, M.D.; Perez-Maqueda, L.A.; Subrt, J.; Criado, J.M. Factors Influencing the Texture and Stability of Maghemite Obtained from the Thermal Decomposition of Lepidocrocite. Chem. Mater. 1999, 11, 1128–1137. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Criado, J.M.; Real, C.; Šubrt, J.; Boháček, J. The use of constant rate thermal analysis (CRTA) for controlling the texture of hematite obtained from the thermal decomposition of goethite. J. Mater. Chem. 1999, 9, 1839–1846. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Sánchez-Jiménez, P.E.; Criado, J.M. Kinetic analysis of solid-state reactions: Precision of the activation energy calculated by integral methods. Int. J. Chem. Kinet. 2005, 37, 658–666. [Google Scholar] [CrossRef] [Green Version]
- Vyazovkin, S.; Burnham, A.K.; Favergeon, L.; Koga, N.; Moukhina, E.; Pérez-Maqueda, L.A.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for analysis of multi-step kinetics. Thermochim. Acta 2020, 689, 178597. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Criado, J.M.; Gotor, F.J.; Málek, J. Advantages of Combined Kinetic Analysis of Experimental Data Obtained under Any Heating Profile. J. Phys. Chem. A 2002, 106, 2862–2868. [Google Scholar] [CrossRef]
- Šesták, J.; Berggren, G. Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures. Thermochim. Acta 1971, 3, 1–12. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Criado, A.J.M.; Sánchez-Jiménez, P.E. Combined Kinetic Analysis of Solid-State Reactions: A Powerful Tool for the Simultaneous Determination of Kinetic Parameters and the Kinetic Model without Previous Assumptions on the Reaction Mechanism. J. Phys. Chem. A 2006, 110, 12456–12462. [Google Scholar] [CrossRef]
- Franco, F.; Cecila, J.; Pérez-Maqueda, L.; Pérez-Rodríguez, J.; Gomes, C. Particle-size reduction of dickite by ultrasound treatments: Effect on the structure, shape and particle-size distribution. Appl. Clay Sci. 2007, 35, 119–127. [Google Scholar] [CrossRef]
- Wiewióra, A.; Pérez-Rodrıguez, J.L.; Perez-Maqueda, L.A.; Drapała, J. Particle size distribution in sonicated high- and low-charge vermiculites. Appl. Clay Sci. 2003, 24, 51–58. [Google Scholar] [CrossRef]
- Perez-Maqueda, L.A.; Blanes, J.M.M.; Pascual, J.; Pérez-Rodríguez, J.L. The influence of sonication on the thermal behavior of muscovite and biotite. J. Eur. Ceram. Soc. 2004, 24, 2793–2801. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Montes, O.M.; González-Macias, E.M.; Franco, F.; Poyato, J.; Pérez-Rodrıguez, J.L. Thermal transformations of sonicated pyrophyllite. Appl. Clay Sci. 2004, 24, 201–207. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.; Criado, J.M.; Perez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Otero-Arean, C.; Letellier, M.; Gerstein, B.C.; Fripiat, J.J. Protonic structure on kaolinite during dehydroxylation studied by proton nuclear magnetic resonance. Summary 1982, 105, 499. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Particle Shape | Meaning of | |||
|---|---|---|---|---|---|
| 2D diffusion | D2 | Cylinder | Base diameter | ||
| 3-D diffusion (Jander) | D3 | Sphere | Diameter | ||
| 3D diffusion (Ginstling–Brounshtein) | D4 | Sphere | Diameter | ||
| 2D interface reaction | R2 | Cylinder | Base diameter | ||
| 3D interface reaction | R3 | Sphere | Diameter |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcenegui-Troya, J.; Sánchez-Jiménez, P.E.; Perejón, A.; Pérez-Maqueda, L.A. Relevance of Particle Size Distribution to Kinetic Analysis: The Case of Thermal Dehydroxylation of Kaolinite. Processes 2021, 9, 1852. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9101852
Arcenegui-Troya J, Sánchez-Jiménez PE, Perejón A, Pérez-Maqueda LA. Relevance of Particle Size Distribution to Kinetic Analysis: The Case of Thermal Dehydroxylation of Kaolinite. Processes. 2021; 9(10):1852. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9101852
Chicago/Turabian StyleArcenegui-Troya, Juan, Pedro E. Sánchez-Jiménez, Antonio Perejón, and Luis A. Pérez-Maqueda. 2021. "Relevance of Particle Size Distribution to Kinetic Analysis: The Case of Thermal Dehydroxylation of Kaolinite" Processes 9, no. 10: 1852. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9101852