
Machine Learning Models for the Classification of CK2 Natural Products Inhibitors with Molecular Fingerprint Descriptors


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
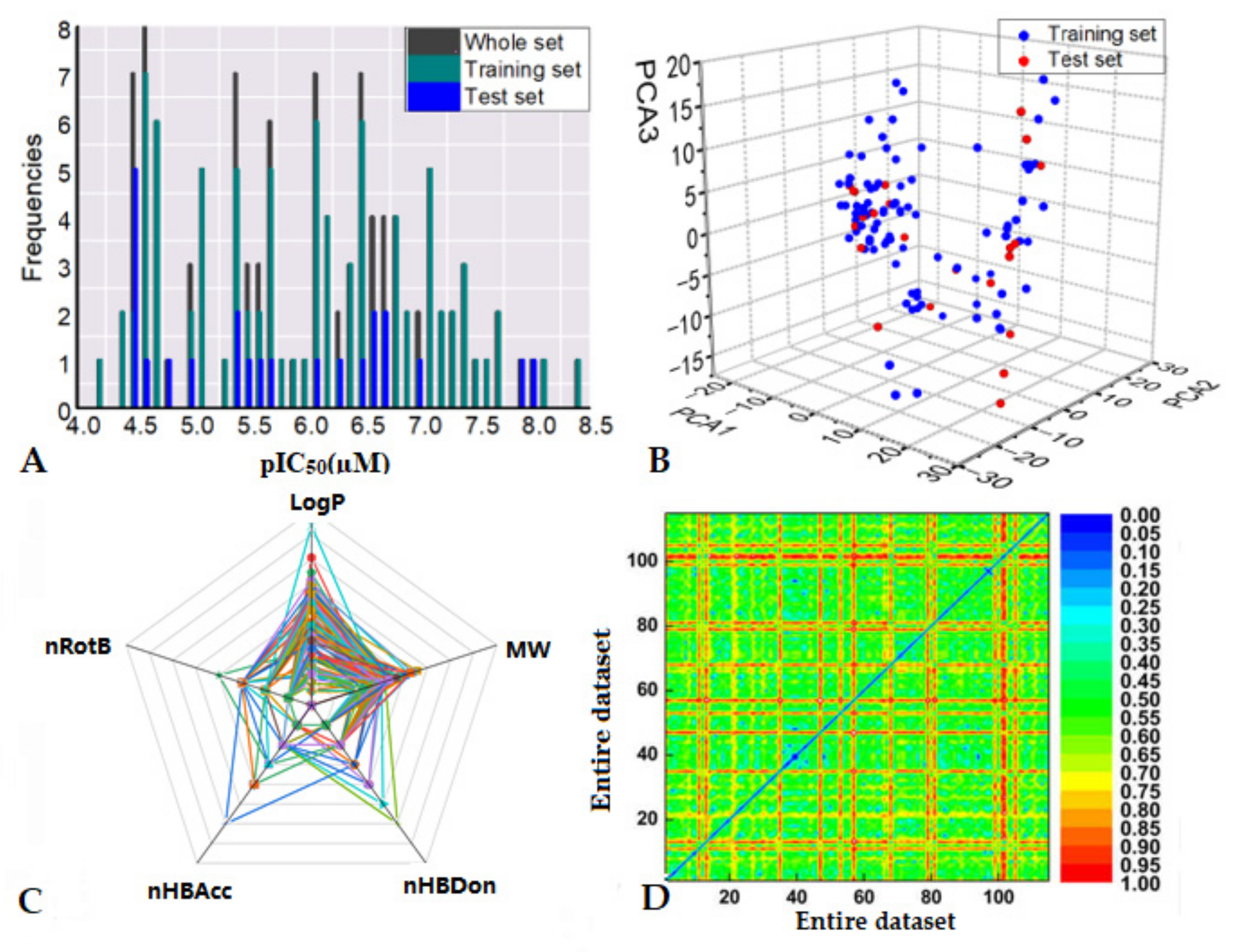
2.1. Data Collection and Chemical Space Distributions
2.2. Molecular Fingerprints and Machine Learning Methods
2.3. Model Performance Evaluation
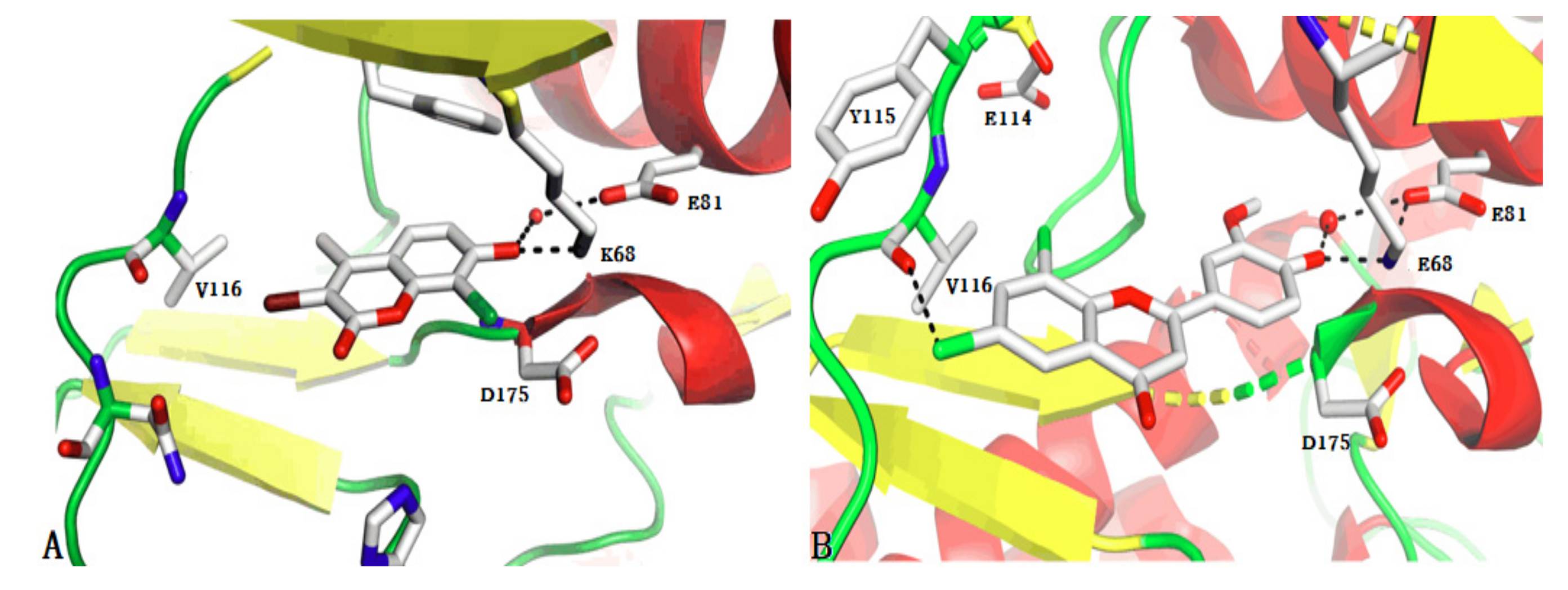
2.4. Identification of Privileged Substructures
3. Results and Discussion
3.1. Dataset Analysis
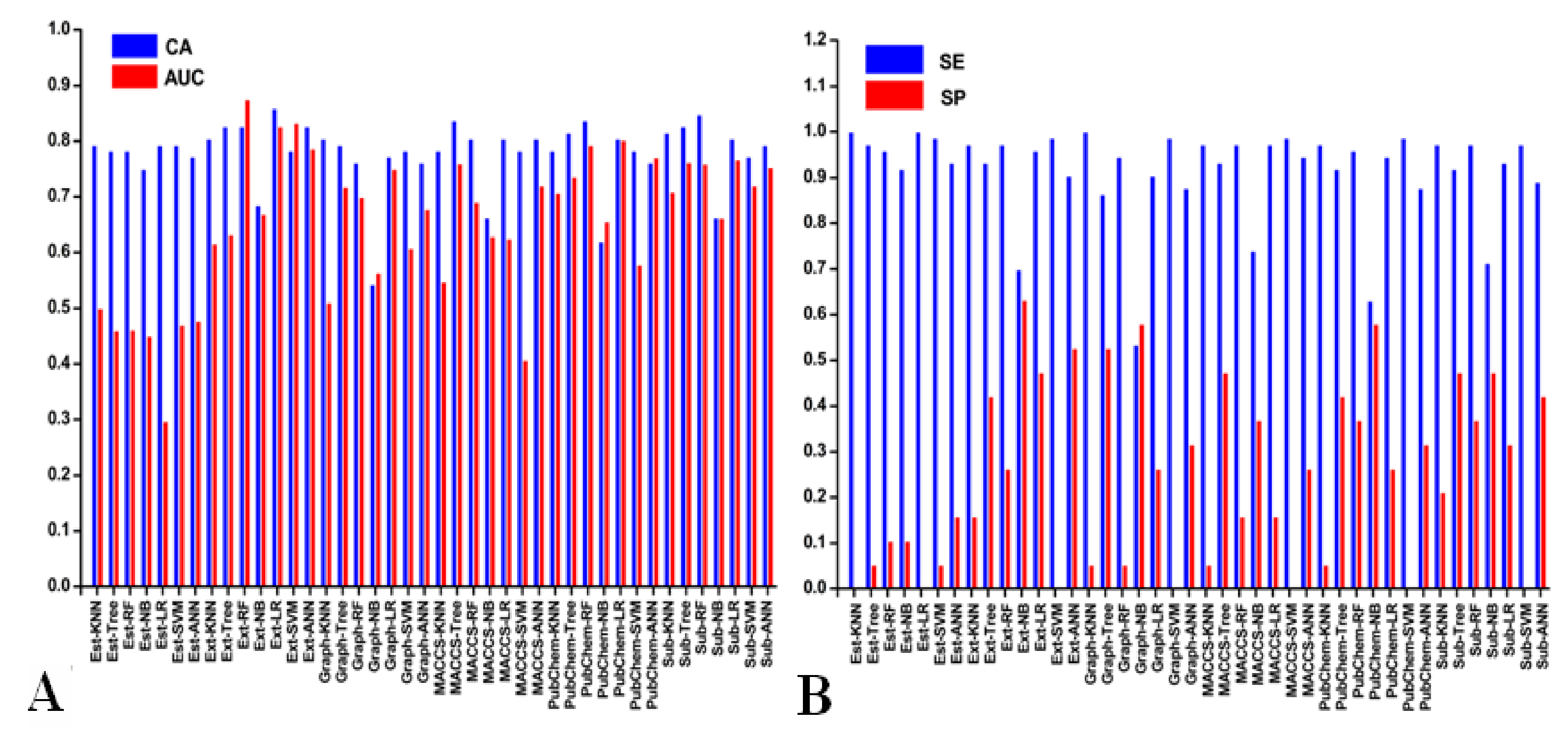
3.2. Performance of 10-Fold Cross-Validation
3.3. Performances of Test Set
3.4. Identification of Privileged Substructures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
References
- Borgo, C.; D’Amore1, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Cesaro, L.; Sarno, S.; Pinna, L.A.; Ruzzene, M.; Salvi, M. How can a traffic light properly work if it is always green? The paradox of CK2 signaling. Crit. Rev. Biochem. Mol. Biol. 2020, 56, 321–359. [Google Scholar]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Marrero, M.C.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020, 182, 685–712. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.E.; Agaisse, H. Casein kinase 2 regulates vaccinia virus actin tail formation. Virology 2012, 423, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Chen, T.; Yang, H.; Chen, Y.; Lin, H.; Qu, W. Small molecule modulators targeting protein kinase CK1 and CK2. Eur. J. Med. Chem. 2019, 181, 111581. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G. The development of CK2 inhibitors: From traditional pharmacology to in silicon rational drug design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Senhwa Biosciences, Inc.Senhwa Biosciences CX-4945 Granted Orphan Drug Designation by the US FDA in Cholangiocarcinoma. Available online: https://www.senhwabio.com/en/news/ec41f1 (accessed on 4 January 2017).
- Cozza, G.; Meggio, F.; Moro, S. The dark side of protein kinase CK2 inhibition. Curr. Med. Chem. 2011, 18, 1884–2867. [Google Scholar] [CrossRef]
- Cozza, G.; Venerando, A.; Sarno, S.; Pinna, L.A. The selectivity of CK2 inhibitorQuinalizarin: A reevaluation. Biol. Andm. Res. Int. 2015, 2015, 734127. [Google Scholar]
- Beutler, J.A. Natural products as a foundation for drug discovery. Curr. Protoc. Pharmacol. 2019, 86, e67. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, B.; Issinger, O.G. Natural compounds and derivatives as Ser/Thr protein kinase modulators and inhibitors. Pharmaceuticals 2019, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Haidar, S.; Jürgens, F.; Aichele, D.; Jose, J. In silico and in vitro studies of natural compounds as human CK2 inhibitors. Curr. Comput.-Aided Drug Des. 2021, 17, 323–331. [Google Scholar] [CrossRef] [PubMed]
- CoMarzec, E.; Świtalska, M.; Winiewska-Szajewska, M.; Wójcik, J.; Wietrzyk, J.; Maciejewska, A.M.; Poznański, J.; Mieczkowski, A. The halogenation of natural flavonoids, baicalein and chrysin, enhances their affinity to human protein kinase CK2. IUBMB Life 2020, 72, 1250–1261. [Google Scholar] [CrossRef]
- Battistutta, R. Protein kinase CK2 in health and disease: Structural bases of protein kinase CK2 inhibition. Cell. Mol. Life Sci. 2009, 66, 1868–1889. [Google Scholar] [CrossRef]
- Ul-Haq, Z.; Ashraf, S.; Bkhaitan, M.M. Molecular dynamics simulations reveal structural insights into inhibitor binding modes and mechanism of casein kinase II inhibitors. J. Biomol. Struct. Dyn. 2019, 37, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhong, R. Docking and 3D-QSAR studies of 7-hydroxycoumarin derivatives as CK2inhibitors. Eur. J. Med. Chem. 2010, 45, 292–297. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, W.; Zhou, Y.; Zhao, H.; Zhong, R. Rational design of Coumarin derivatives as CK2 inhibitors by improving the interaction with the hinge region. Mol. Inform. 2016, 35, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Di-wu, L.; Li, L.L.; Wang, W.J.; Xie, H.Z.; Yang, J.; Zhang, C.H.; Zhong, L.; Feng, S.; Yang, S.Y. Identification of CK2 inhibitors with new scaffolds by a hybrid virtual screening approach based on Bayesian model; pharmacophore hypothesis and molecular docking. J. Mol. Graph. Model. 2012, 36, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Tutone, M.; Pibiri, I.; Perriera, R.; Campofelice, A.; Culletta, G.; Melfi, R.; Pace, A.; Almerico, A.M.; Lentini, L. Pharmacophore-based design of new chemical scaffolds as translational read though-inducing drugs (TRIDs). ACS Med. Chem. Lett. 2020, 11, 747–753. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, W.; Zhang, D.; Wang, S.; Shi, J.; Niu, Y. Recent advances in the machine learning-based drug-target interaction prediction. Curr. Drug Metab. 2019, 20, 194–202. [Google Scholar] [CrossRef]
- Pena-Guerrero, J.; Nguewa, P.A.; Garcia-Sosa, A.T. Machine learning, artificial intelligence, and data science breaking into drug design and neglected diseases. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2021, 11, e1513. [Google Scholar] [CrossRef]
- Yang, M.; Tao, B.; Chen, C.; Jia, W.; Sun, S.; Zhang, T. Machine learning models based on molecular fingerprints and an extreme gradient boosting method lead to the discovery of JAK2 inhibitors. J. Chem. Inf. Model. 2019, 59, 5002–5012. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pérez, R.; Miljković, F.; Bajorath, J. Assessing the information content of structural and protein-ligand interaction representations for the classification of kinase inhibitor binding modes via machine learning and active learning. J. Cheminf. 2017, 12, 362020. [Google Scholar] [CrossRef]
- Chilin, A.; Battistutta, R.; Bortolato, A. Coumarin as attractive casein kinase2 (CK2) inhibitor scaffold: An integrate approach to elucidate the putative binding motif and explain structure-activity relationships. J. Med. Chem. 2008, 51, 752–759. [Google Scholar] [CrossRef]
- Golub, A.G.; Bdzhola, V.G.; Ostrynska, O.V.; Kyshenia, I.V.; Sapelkin, V.M.; Prykhod’ko, A.O. Discovery and characterization of synthetic 4′-hydroxyflavones New CK2 inhibitors from flavone family. Bioorgan. Med. Chem. 2013, 21, 6681–6689. [Google Scholar] [CrossRef]
- DeMoliner, E.; Moro, S.; Sarno, S.; Zagotto, G.; Zanotti, G.; Pinna, L.A. Inhibition of protein kinase CK2 by anthraquinone-related compounds. J. Biol. Chem. 2003, 278, 1831–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozza, G.; Zonta, F.; DalleVedove, A.; Venerando, A.; Dall’Acqua, S.; Battistutta, R.; Ruzzene, M. Biochemical and cellular mechanism of protein kinase CK2 inhibition by deceptive curcumin. FEBS J. 2020, 287, 1850–1864. [Google Scholar] [CrossRef]
- Dragon Software for Molecular Descriptor Calculation V7.0.6, KodeSrl. Available online: https://chm.kode-solutions.net/ (accessed on 3 September 2017).
- Heikamp, K.; Bajorat, J. Fingerprint design and engineering strategies: Rationalizing and improving similarity search performance. Future Med. Chem. 2012, 4, 1945–1959. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2010, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.M.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Dreiseitl, S.; Ohno-Machado, L. Logistic regression and artificial neural network classification models: A methodology review. J. Biomed. Inform. 2002, 35, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Watson, P. Naive Bayes classification using 2D pharmacophore feature triplet vectors. J. Chem. Inf. Model. 2008, 48, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Basheer, I.A.; Hajmeer, M. Artificial neural networks: Fundamentals, computing, design, and application. J. Microbiol. Methods 2000, 43, 3–31. [Google Scholar] [CrossRef]
- Bouboulis, P.; Theodoridis, S.; Mavroforakis, C.; Dalla, L. Complex support vector machines for regression and quaternary classification. IEEE Trans. Neural Netw. Learn. Syst. 2014, 26, 1260–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.; Sheridan, R.; Feuston, B. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Plewczynski, D.; Spieser, S.; Koch, U. Assessing different classification methods for virtual screening. J. Chem. Inf. Model. 2006, 46, 1098–1106. [Google Scholar] [CrossRef]
- Fan, T.; Sun, G.; Zhao, L.; Cui, X.; Zhong, R. QSAR and classification study on prediction of acute oral toxicity of N-nitroso compounds. Int. J. Mol. Sci. 2019, 19, 3015. [Google Scholar] [CrossRef] [Green Version]
- Perez-Garrido, A.; Helguera, A.M.; Borges, F.; Cordeiro, M.; Rivero, V.; Escudero, A.G. Two new parameters based on distances in a receiver operating characteristic chart for the selection of classification models. J. Chem. Inf. Model. 2011, 51, 2746–2759. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cheng, F.X.; Xu, Y.; Li, W.H.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Sun, G.L.; Fan, T.; Sun, X.; Hao, Y.; Cui, X.; Zhao, L.; Ren, T.; Zhou, Y.; Zhong, R.; Peng, Y. In Silico Prediction of O⁶-Methylguanine-DNA Methyltransferase inhibitory potency of base analogs with QSAR and machine learning methods. Molecules 2018, 23, 2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiguchi, Y.; Nakaniwa, T.; Kinoshita, T.; Nakanishi, I.; Kitaura, K.; Hirasawa, A. Structural insight into human CK2α in complex with the potent inhibitor ellagic acid. Bioorgan. Med. Chem. 2009, 19, 2920–2923. [Google Scholar] [CrossRef] [PubMed]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12 PubChem: Integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Sawada, R.; Kotera, M.; Yamanishi, Y. Benchmarking a wide range of chemical descriptors for drug-target interaction prediction using a chemogenomic approach. Mol. Inform. 2014, 33, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, X.; Zhang, N.; Zhong, R. Structural basis for low-affinity binding of non-R2 carboxylate-substituted tricyclic quinoline analogs to CK2α: Comparative molecular dynamics simulation studies. Chem. Biol. Drug Des. 2015, 85, 189–200. [Google Scholar] [CrossRef]
- Shinada, N.K.; Brevern, A.G.; Schmidtke, P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, N.; Zhao, L.; Hu, L.; Cortopassi, W.A.; Jacobson, M.P.; Li, X.; Zhong, R. Structure-based identification of novel CK2 inhibitors with a linear 2-propenone scaffold as anti-cancer agents. Biochem. Biophys. Res. Commun. 2019, 512, 208–212. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Data Set | Model | CA | AUC | SE | SP | PP | NP | MCC | TP | TN | FP | FN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Training set | Ext-RF | 0.826 | 0.875 | 0.97 | 0.26 | 0.835 | 0.714 | 0.360 | 71 | 5 | 14 | 2 |
| Ext-SVM | 0.830 | 0.833 | 0.99 | 0.00 | 0.791 | 0 | −0.050 | 72 | 0 | 19 | 1 | |
| Ext-LR | 0.859 | 0.826 | 0.96 | 0.47 | 0.875 | 0.750 | 0.520 | 70 | 9 | 10 | 3 | |
| PubChem-LR | 0.804 | 0.802 | 0.95 | 0.26 | 0.831 | 0.556 | 0.283 | 69 | 5 | 14 | 4 | |
| PubChem-RF | 0.837 | 0.793 | 0.96 | 0.37 | 0.853 | 0.700 | 0.426 | 70 | 7 | 12 | 3 | |
| Ext-ANN | 0.826 | 0.787 | 0.90 | 0.53 | 0.880 | 0.589 | 0.449 | 66 | 10 | 9 | 7 | |
| PubChem-ANN | 0.761 | 0.771 | 0.88 | 0.32 | 0.831 | 0.400 | 0.210 | 64 | 6 | 13 | 9 | |
| Sub-LR | 0.804 | 0.767 | 0.93 | 0.32 | 0.840 | 0.545 | 0.309 | 68 | 6 | 13 | 5 | |
| Sub-Tree | 0.826 | 0.762 | 0.92 | 0.47 | 0.870 | 0.600 | 0.430 | 67 | 9 | 10 | 6 | |
| MACCS-Tree | 0.837 | 0.760 | 0.93 | 0.47 | 0.871 | 0.643 | 0.457 | 68 | 9 | 10 | 5 | |
| Test set | Ext-RF | 0.739 | 0.850 | 1.00 | 0.25 | 0.714 | 1.00 | 0.422 | 15 | 2 | 6 | 0 |
| Ext-SVM | 0.652 | 0.850 | 1.00 | 0.00 | 0.652 | 0 | 0 | 15 | 0 | 8 | 0 | |
| Ext-LR | 0.826 | 0.800 | 1.00 | 0.50 | 0.789 | 1.00 | 0.628 | 15 | 4 | 4 | 0 | |
| PubChem-LR | 0.783 | 0.833 | 1.00 | 0.38 | 0.750 | 1.00 | 0.530 | 15 | 3 | 5 | 0 | |
| PubChem-RF | 0.739 | 0.875 | 0.93 | 0.38 | 0.737 | 0.750 | 0.387 | 14 | 3 | 5 | 1 | |
| Ext-ANN | 0.826 | 0.758 | 1.00 | 0.50 | 0.789 | 1.00 | 0.628 | 15 | 4 | 4 | 0 | |
| PubChem-ANN | 0.826 | 0.933 | 1.00 | 0.50 | 0.789 | 1.00 | 0.628 | 15 | 4 | 4 | 0 | |
| Sub-LR | 0.783 | 0.675 | 0.87 | 0.63 | 0.812 | 0.714 | 0.509 | 13 | 5 | 3 | 2 | |
| Sub-Tree | 0.739 | 0.654 | 0.93 | 0.38 | 0.737 | 0.750 | 0.387 | 14 | 3 | 5 | 1 | |
| MACCS-Tree | 0.739 | 0.783 | 0.93 | 0.38 | 0.737 | 0.750 | 0.387 | 14 | 3 | 5 | 1 |
| No. | Privileged Substructures | General Substructures | Representative Compounds | IG | FP | FN |
|---|---|---|---|---|---|---|
| PubChemFP439 | C(-C)(-N)(=O) |  |  | 0.007 | 1.31(2) | 0(0) |
| PubChemFP807 | OC1CC(Br)CCC1 |  |  | 0.010 | 1.31(3) | 0(0) |
| PubChemFP38 PubChemFP815 | >= 2 ClClC1CC(Cl)CCC1 |  |  | 0.014 0.014 | 1.31(4) 1.31(4) | 0(0) 0(0) |
| PubChemFP193 | >= 3 saturated or aromatic carbon-only ring size 6 |  |  | 0.014 | 1.31(4) | 0(0) |
| PubChemFP806 | OC1CC(Cl)CCC1 |  |  | 0.014 | 1.31(4) | 0(0) |
| PubChemFP785 | OC1CCC(Cl)CC1 |  |  | 0.032 | 1.31(9) | 0(0) |
| PubChemFP818 | CC1C(C)CCCC1 |  |  | 0.032 | 1.31(9) | 0(0) |
| PubChemFP505 PubChemFP551 PubChemFP827 | Cl-C:C-O Cl-C-C-O OC1C(Cl)CCCC1 |  |  | 0.011 0.011 0.011 | 1.19(10) 1.19(10) 1.19(10) | 0.39(1) 0.39(1) 0.39(1) |
| PubChemFP801 | CC1CC(Cl)CCC1 |  |  | 0.027 | 1.23(16) | 0.24(1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Bi, M.; Zhang, X.; Zhang, N.; Sun, G.; Zhou, Y.; Zhao, L.; Zhong, R. Machine Learning Models for the Classification of CK2 Natural Products Inhibitors with Molecular Fingerprint Descriptors. Processes 2021, 9, 2074. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112074
Liu Y, Bi M, Zhang X, Zhang N, Sun G, Zhou Y, Zhao L, Zhong R. Machine Learning Models for the Classification of CK2 Natural Products Inhibitors with Molecular Fingerprint Descriptors. Processes. 2021; 9(11):2074. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112074
Chicago/Turabian StyleLiu, Yuting, Mengzhou Bi, Xuewen Zhang, Na Zhang, Guohui Sun, Yue Zhou, Lijiao Zhao, and Rugang Zhong. 2021. "Machine Learning Models for the Classification of CK2 Natural Products Inhibitors with Molecular Fingerprint Descriptors" Processes 9, no. 11: 2074. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112074