Requiem for the Rate-Determining Step in Complex Heterogeneous Catalytic Reactions?


Process Chemistry Centre, Åbo Akademi University, 20500 Turku/Åbo, Finland
Reactions 2020, 1(1), 37-46; https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1010004
Submission received: 1 September 2020
/
Revised: 8 September 2020
/
Accepted: 9 September 2020
/
Published: 10 September 2020
(This article belongs to the Special Issue Mechanism and Kinetics of Complex Reactions— 2020 Selected Papers from Reactions’ Editorial Board Members)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The concept of the rate determining step, i.e., the step having the strongest influence on the reaction rate or even being the only one present in the rate equation, is often used in heterogeneous catalytic reactions. The utilization of this concept mainly stems from a need to reduce complexity in deriving explicit rate equations or searching for a better catalyst based on the theoretical insight. When the aim is to derive a rate equation with eventual kinetic modelling for single-route mechanisms with linear sequences, the analytical rate expressions can be obtained based on the theory of complex reactions. For such mechanisms, a single rate limiting step might not be present at all and the common practice of introducing such steps is due mainly to the convenience of using simpler expressions. For mechanisms with a combination of linear and nonlinear steps or those just comprising non-linear steps, the reaction rates are influenced by several steps depending on reaction conditions, thus a reduction in complexity to a single rate limiting step can lead to misinterpretations. More widespread utilization of a microkinetic approach when the reaction rate constants can be computed with reasonable accuracy based on the theoretical insight, and availability of software for kinetic modelling, when a system of differential equations for reactants and products will be solved together with differential equations for catalytic species and the algebraic conservation equation for the latter, will eventually make the concept of the rate limiting step obsolete.
1. Introduction
The kinetics of heterogeneous catalytic reactions has been the focus of numerous studies because of its theoretical and practical importance [1,2,3,4,5,6,7,8,9,10]. Kinetic analysis was applied for decades to assist reactor design and scaling up of various reactions relevant for oil refining and synthesis of basis and specialty chemicals. More recently, because of a widespread application of so-called flow chemistry to streamline pharmaceutical research and products, kinetic analysis also started to be more widely utilized in the field of fine chemicals [11,12,13]. It was argued [11] that the framework of a mechanistically based rate equation combined with in situ experimental data is useful for the interpretation of complex reaction networks. Increasing complexity of the catalytic reactions, either relevant for oil refining or pharmaceutical synthesis, poses some restrictions regarding incorporation of such complex mechanisms into tractable rate equations.
Thus, another trend is to simplify complex reaction networks and assume that there is a limiting step, the rate of which is a good approximation of the reaction rate of the whole network [14,15]. This approach of a rate-determining step (RDS) became very popular in chemical reaction engineering, allowing the derivation of kinetic equations for reaction mechanisms of differing complexity. Such an assumption of a rate-determining step makes the rate expressions more tractable, facilitating the kinetic modelling of mainly steady-state kinetics even at the expense of losing important mechanistic information. Moreover, in several instances, without a substantial reduction in complexity, kinetic modelling can be computationally very demanding, especially when coupled with transport phenomena [16].
On the other hand, the concept of RDS was suggested to give fruitful ideas for modifying the reaction conditions or design the catalyst. Subsequently, there have been many theoretical papers addressing how to determine the step which is the rate determining [17,18]. The most popular is probably the degree of rate control proposed by Campbell, based on the general principles of differential sensitivity analysis to determine sensitivity of the output to one input parameter [19,20,21,22,23,24,25]. It was argued [16] that an approach analogous to electrical networks is even better suited for the identification of the key steps determining the reaction rate allowing the derivation of accurate rate expressions even for mechanisms with nonlinear steps. Such nonlinear steps are present not only in heterogeneous catalytic reactions, such as, for example, a water gas shift reaction [16], decomposition of ammonia [26,27] or methanol [28], but also in organometallic catalysis. In the latter case, the reaction order in catalysts deviates from unity [5]. The formation of dimeric catalytic species, on the contrary, results in the reaction order being below one, which is rather rare [29,30,31].
Contrary to the case of linear (Christiansen) sequences of a single-route reaction [5,32,33] for single-route mechanisms with nonlinear steps, a general expression for the reaction rate in steady-state and quasi-steady-state conditions cannot be derived [34]. When such derivation is possible, the rate expressions become rather complicated to handle [5,35,36,37].
To overcome difficulties in deriving explicit rate expressions, a semi-empirical kinetic polynomial was proposed [38] as a substitute of analytical rate equations.
However, in catalysis by solids or organometallic complexes, quite often either there are nonlinear steps involved or the rate equations for even single-route mechanisms and obviously multi route reactions do not have rate limiting steps [34].
It was recognized a long time ago that for multi-route (or composite) reactions, the concept of the rate-controlling steps is not required [34,39], while in other cases it should be considered with care, as misinterpretation is easily possible.
In the current work, several cases of complex single- and multi-route heterogeneous catalytic reactions will be presented, highlighting an apparent danger of falling into a trap of using the rate determining step, which leads to confusion rather than clarification of reaction mechanisms.
2. Three Step Sequence
Let us consider first a three-step linear sequence (Figure 1)
The general equation for this three-step sequence in the case of all reversible steps can be written in a form of frequencies of steps [6]
where r is the reaction rate, is the frequency of step 1, etc., and Ccat is the catalyst concentration. The frequencies of steps are obtained by dividing the rate expressions of a particular step by the concentration of catalytic species in that expression. For example, in the case of the reaction mechanism presented in Figure 2, the general form of the Equation (1) gives [6]
Thus, the frequency of the first step in the forward direction is , while for the reverse step it would be just . When all steps are irreversible, Equation (2) can be easily simplified, giving
It follows from Equation (3) that depending on the values of partial pressures of, for example, hydrogen, the reaction order can change from unity at low pressures, to zero at high hydrogen pressures. The relative contribution of the last term in the denominator of Equation (3) in comparison with two other terms defines the reaction order towards ethylene.
This example illustrates an apparent danger of defining the rate and even more turnover frequency through just one rate limiting step without taking into account kinetic regularities. To highlight the need to consider all steps in a linear catalytic sequence even further, let us consider the apparent activation energy for the three-step mechanism (Equation (3)). In what follows, the term “apparent” corresponds to the observed activation energy of a complex multistep reaction in the kinetic regime, which should not be confused with the case when the observed kinetics is influenced by the transport phenomena.
A more simple case of the two-step sequence was addressed in the literature [41] using the definition of the apparent activation energy
and giving
As follows from Equation (5), the apparent activation energy depends not only on the step with the highest activation energy, but also on the contribution of the other step with a lower activation energy. For the three-step sequence using Equations (3) and (4), one gets
Some terms in Equation (6) can be easily differentiated
Taking Equation (7) into account and also using a standard approach for the differentiation of a complex function
one gets the final expression for the activation energy of a three step sequence
It can be shown that in only exceptional cases the apparent activation energy of the overall reaction is determined by the activation energy of only one rate-limiting step in a steady-state sequence.
3. Four Step Sequence
As an example of such a sequence, the liquid-phase hydrogenation of aromatic compounds will be considered. The reaction kinetics was extensively investigated previously in [42,43,44,45,46]. The following generic scheme for hydrogenation of an aromatic compound A to the product B was proposed
where Y is cyclohexene or its derivative, AH2 and AH4 are intermediate complexes, step 3′ is fast because hydrogenation of cycloalkenes is much faster than aromatic compounds and step 4 is at quasi-equilibrium.
1.*A + H2 ⇔ *AH2
2.*AH2 + H2 ⇔ *AH4
3.*AH4 ⇒ *Y
3′.*Y + H2 ⇔ *B (fast)
4.*B + A Ξ *A + B
A + 3H2 = B
2.*AH2 + H2 ⇔ *AH4
3.*AH4 ⇒ *Y
3′.*Y + H2 ⇔ *B (fast)
4.*B + A Ξ *A + B
A + 3H2 = B
Because of irreversibility of step (3), Equations (11) and (12) can be simplified
Step 4 is at quasi-equilibrium, and therefore the rates are fast in both directions. Further simplifications of Equation (13) are thus possible by dividing the numerator and the denominator by , neglecting subsequently the terms and . Then the following holds
Replacing frequencies in Equation (15) with the explicit expressions, one gets
where . For a binary mixture of the reactant and substrate, it is possible to use mole fractions instead of concentrations. Moreover, quite often partial pressures of hydrogen are applied instead of concentrations, resulting in another form of the rate equation
where is the mole fraction of the reactant. Equation (16) contains the modified rate constants as instead of hydrogen concentration in the liquid phase, the pressure of hydrogen is used.
Equation (17) presented in a slightly different form
was derived in [43] using a more tedious procedure, namely applying directly the steady-state conditions
as well as the quasi-equilibrium for step 4 and finally the balance equation (i.e., the sum of all coverages is equal to unity).
All steps in Equation (10) are needed to explain the observed kinetic regularities, because it was demonstrated that at low hydrogen partial pressures, the reaction order in hydrogen can exceed unity, which is possible when k−1 ≠ 0, while at high pressures the reaction kinetics obeys frequently observed zero orders in the substrate and hydrogen. In the latter case, the overall rate is determined by the isomerization of the adsorbed AH4 species. The surface of the catalyst is completely covered with this complex and the reaction rate ceases to depend on either hydrogen pressure or the concentration of the substrate.
Apparently, the concept of a single rate-determining step is not able to account for a rich kinetic behavior, observed experimentally in hydrogenation of aromatics.
4. Oxidative Dehydrogenation of Ethanol: A Multi-Route Mechanism with Nonlinear Steps
The mechanism and kinetics of oxidative dehydrogenation of ethanol over gold catalysts was recently reported [47]. DFT calculations indicated that the activation of molecular oxygen is facilitated in the presence of ethanol acting as a hydrogen donor. As a result of hydrogen abstraction from the donor leading to the formation of an OOH intermediate, the dioxygen undergoes dissociation on the catalyst surface. The scheme of the reaction mechanism is given below
In (20), on the right hand side of the equations for the steps (i.e., 1 to 7c), the stoichiometric numbers (i.e., 0, 1, 2) along alternative routes N(1a) - N(1c) are given [32]. These numbers can be equal to zero, if a particular step is not involved in the reaction mechanism. In Equation (20), EtOH stands for ethanol, while AcH represents acetaldehyde and * denotes a surface site.
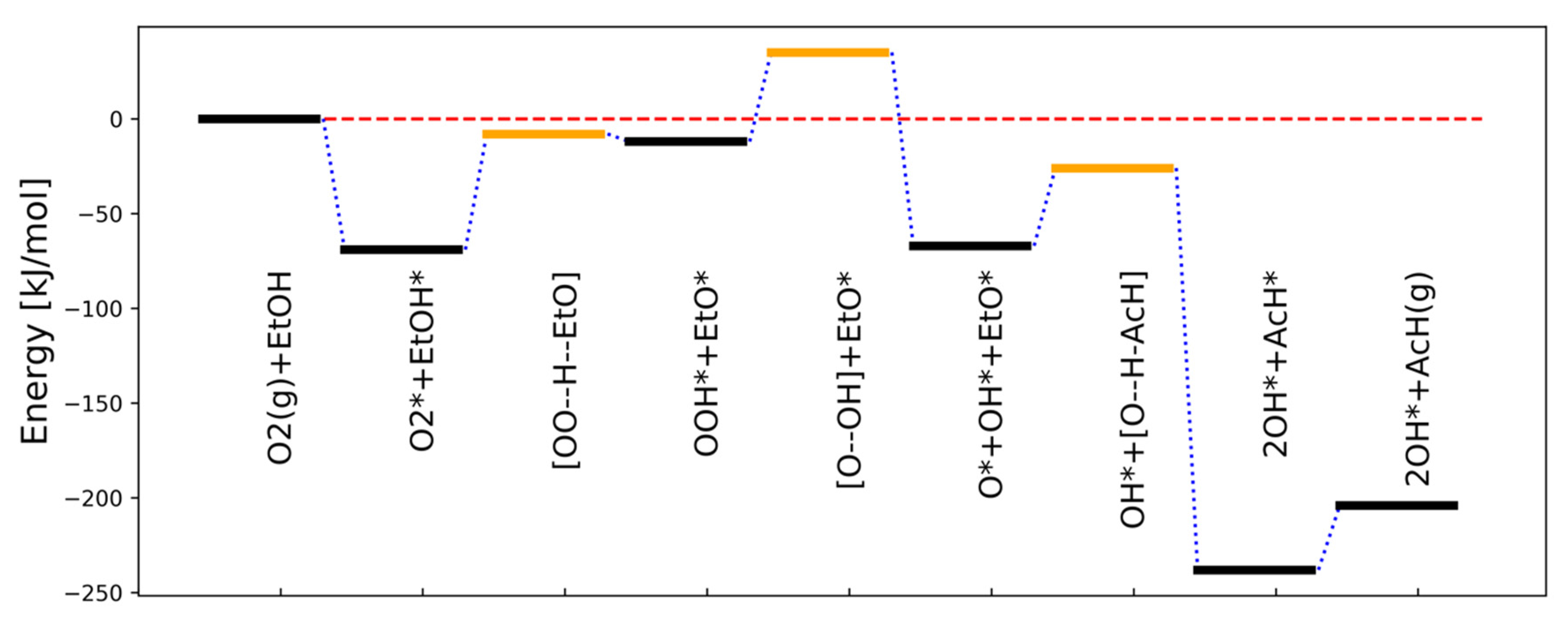
According to the DFT calculations [47] illustrated in Figure 3, the largest activation barrier corresponds to step 3 in Equation (20), and thus superficially it can be considered in the conventional approaches as the rate limiting step. However, the activation energy of the same step in the reverse direction is very low and step 3 can even be viewed to be close to the quasi-equilibrium.
From the considerations above on the apparent activation energy of the three-step sequence with linear steps, it is clear that the apparent activation energy and the reaction rate depend on the contribution of all steps in the mechanism, which could be treated using the steady-state approximation. This approach is possible because of the specific features of the reaction mechanism comprising only nonlinear steps and thus allowing derivation of an analytic rate expression. Such derivation was done in [47] based on the DFT computed reaction energies assuming quasi-equilibria for steps 1 and 2, irreversibility for steps 4 and 5 and reversibility for steps 3 and 6.
From the viewpoint of the global reaction kinetics, not only steps 3–6 are important but also the final step in the mechanism, step 7, reflecting the fate of atomically adsorbed oxygen. Therefore, three options are possibly influencing the stoichiometric numbers of other steps. Step 7a in the route N(1a) corresponds to recombination of atomic oxygen, while such recombination in step 7c (route N(1c)) involves a reaction with a peroxy species OOH. In the route N(1b), atomically adsorbed oxygen assists in the abstraction of hydrogen from the substrate.
Derivation of the rate expression (per mole of oxygen) for the route N(1b) was described in [47], giving
Just for illustration purposes, let us consider a case when step 3 is at quasi-equilibrium and step 6 is irreversible. Equation (21) is then transformed to
For two other options also considering the quasi-equilibria of the step 3, the rates are for the routes N(1b)
and N(1a), respectively
illustrating that the rate expression and even the kinetic regularities which follow from the corresponding rate expressions are very sensitive to the chemistry of step 7 and the rate constant of this step. The mechanism N(1a) is less probable compared to other alternatives considering a high activation barrier anticipated for this step [47], while other options in principle are possible. For kinetic modelling in [47], a feasible channel of atomically adsorbed oxygen consumption with a low activation energy barrier was considered to be step 7b (Equation (20)).
5. Conclusions and Outlook
Apparently, the concept of the rate limiting will continue to be utilized in the future. This concept is useful as a part of the education of future specialists in chemical reaction engineering and catalyst development. For simple single-route reaction mechanisms, the application of the rate-determining step along with the quasi-equilibria of other steps results in easily derived rate equations. For more complex cases with several linear steps, the general approach based on the theory of complex reactions of Horiuti–Temkin allows the derivation of the corresponding rate equations. Apparent difficulties in doing this, as illustrated above for the four-step sequence, often result in a reductionistic approach assuming a rate-determining step either based on chemical intuition or a more rigorous theoretical analysis based on quantum chemical calculations.
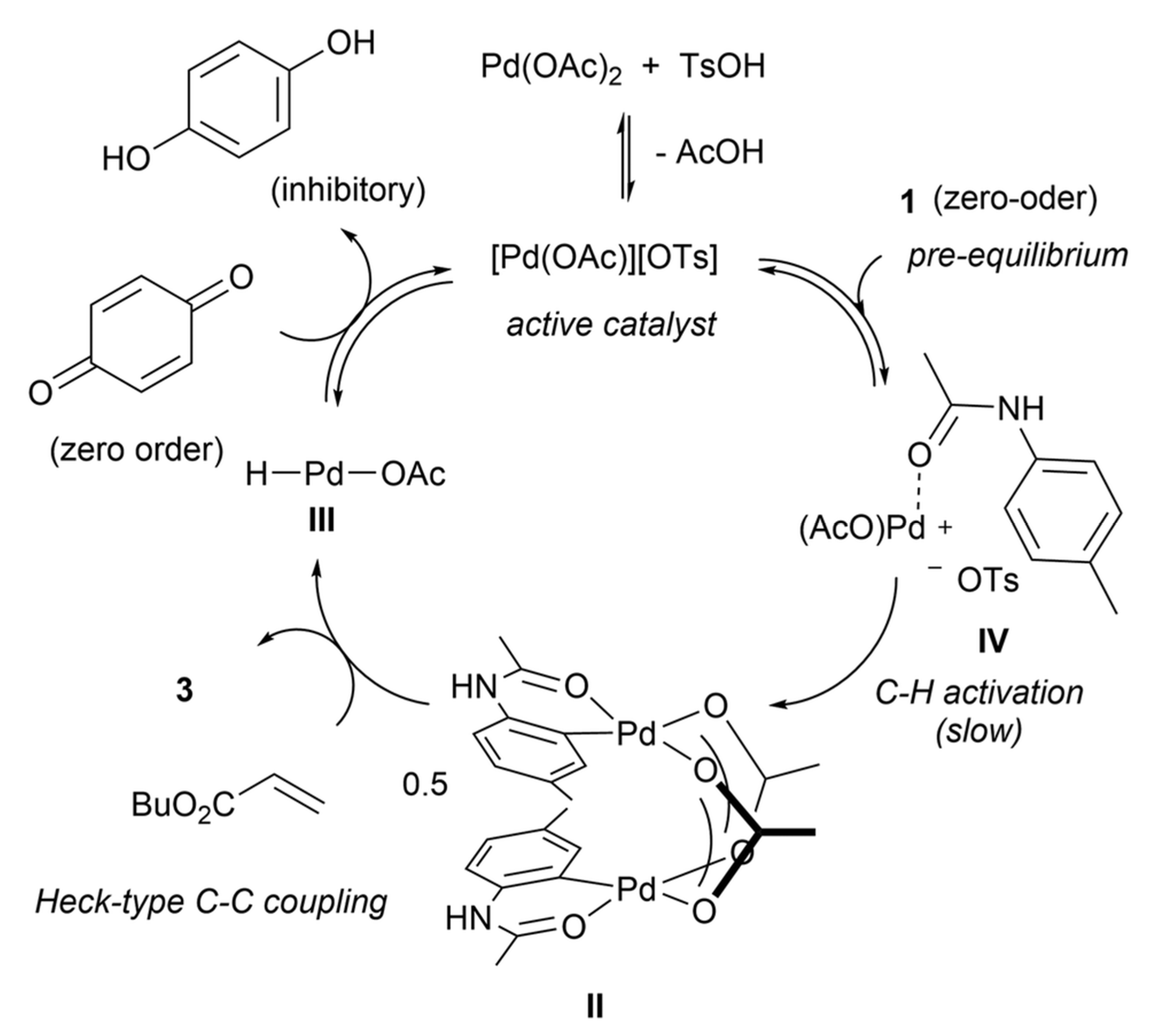
There are cases when the rate equation for linear sequences with a small number of steps was not derived and a system of differential equations was solved even if the rate equation for the steady -state system can be readily derived. This was done, for example in [48], when a solver of differential equations was used for a four step sequence of the Fujiwara–Moritani reaction (Figure 4), unfortunately not reporting how the balance equation for catalytic species was taken into account,
For a particular case of the mechanism in Figure 4, the rate equation can be easily derived as this mechanism is a special case of Equations (11) and (12) with irreversible steps 2 and 3 simply resulting in
Or, after introducing the explicit expressions for the frequencies of steps
In a more general case, when the reaction mechanisms comprise linear and nonlinear steps without any rate-limiting ones, derivation of an explicit rate equation can be too tedious. Instead, a comparison between the experimental and calculations should be done in an implicit form using a system of differential equations considering also an algebraic balance equation for the catalytic species. Availability of computer programs with a customer written subroutine comprising a set of elementary reactions will pave the way for eventual marginalization of the concept of the rate-determining step in the kinetic modelling of complex heterogeneous catalytic reactions.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Tsukamoto, M.; Gopalaiah, K.; Kagan, H.B. Equilibrium of homochiral oligomerization of a mixture of enantiomers. Its relevance to nonlinear effects in asymmetric catalysis. Phys. Chem. 2008, 112, 15361–15368. [Google Scholar] [CrossRef]
- Blackmond, D.G. Kinetic profiling of catalytic organic reactions as a mechanistic tool. Am. J. Chem. Soc. 2015, 137, 10852–10866. [Google Scholar] [CrossRef]
- Helfferich, F.G. Kinetics of Homogeneous Multistep Reactions. In Comprehensive Chemical Kinetics; Compton, R.G., Hancock, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 38. [Google Scholar]
- Leskovac, V. Comprehensive Enzyme Kinetics; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2003. [Google Scholar]
- Temkin, O.N. Homogeneous Catalysis with Metal Complexes: Kinetic Aspects and Mechanisms; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Murzin, D.Y.; Salmi, T. Catalytic Kinetics, Chemistry and Engineering, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Salmi, T.O.; Mikkola, J.-P.; Wärnå, J. Chemical Reaction Engineering and Reactor Technology; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Fogler, H.S. Elements of Chemical Reaction Engineering, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 1998. [Google Scholar]
- Marin, G.; Yhablonsky, G.S. Kinetics of Chemical Reactions; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Kapteijn, F.; Berger, R.J.; Moulijn, J.A. Rate Procurement and Kinetic Modelling, Handbook of Heterogeneous Catalysis; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Blackmond, D.G. Requiem for the reaction rate equation? Catal. Lett. 2002, 83, 133–136. [Google Scholar] [CrossRef]
- Ball, L.T.; Corrie, T.J.A.; Cresswell, A.J.; Lloyd-Jones, G.C. Kinetic analysis of domino catalysis: A case study on gold-catalyzed arylation. ACS Catal. 2020. [Google Scholar] [CrossRef]
- Schmidt, O.P.; Dechert, A.-M.; Garnsey, M.; Wisniewska, H.; Blackmond, D.G. Kinetic analysis of catalytic organic reactions using a temperature scanning protocol. ChemCatChem 2019, 11, 3808–3813. [Google Scholar] [CrossRef] [Green Version]
- Gorban, A. Model reduction in chemical dynamics: Slow invariant manifolds, singular perturbations, thermodynamic estimates, and analysis of reaction graph. arXiv 2018, arXiv:1802.05745. [Google Scholar] [CrossRef] [Green Version]
- Constales, D.; Yablonsky, G.S.; D’Hooge, D.R.; Thybaut, J.W.; Marin, G.B. Physicochemical Principles of Simplifications of Complex Models, Advanced Data Analysis and Modelling in Chemical Engineering; Springer Science+Business Media: Berlin, Germany, 2017; Chapter 4; pp. 83–103. [Google Scholar]
- O’Malley, P.D.; Datta, R.; Vilekar, S.A. Ockham’s razor for paring microkinetic mechanisms: Electric analogy vs. Campbell’s degree of rate control. AIChE J. 2015, 61, 4332–4346. [Google Scholar] [CrossRef]
- Motagamwala, A.H.; Dumesic, J.A. Analysis of reaction schemes using maximum rates of constituent steps. Proc. Natl. Acad. Sci. USA 2016, 113, E2879–E2888. [Google Scholar] [CrossRef] [Green Version]
- Motagamwala, A.H.; Ball, M.R.; Dumesic, J.A. Microkinetic analysis and scaling relations for catalyst design. Ann. Rev. Chem. Biomol. Eng. 2018, 9, 413–450. [Google Scholar] [CrossRef]
- Steigelmann, C.; Andreasen, A.; Campbell, C.T. Degree of rate control: How much the energies of intermediates and transition states control rates. JACS 2009, 131, 8077–8082. [Google Scholar] [CrossRef]
- Campbell, C.T. The degree of rate control: A powerful tool for catalysis research. ACS Catal. 2017, 7, 2770–2779. [Google Scholar] [CrossRef] [Green Version]
- Wolcott, C.A.; Medford, A.J.; Studt, F.; Campbell, C.T. Degree of rate control approach to computation catalyst screening. J. Catal. 2015, 330, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Campbell, C.T. Apparent activation energies in complex reaction mechanisms: A simple relationship via degrees of rate control. ACS Catal. 2019, 9, 9465–9473. [Google Scholar] [CrossRef]
- Mao, Z.; Campbell, C.T. Kinetic isotope effects: Interpretation and prediction using degrees of rate control. ACS Catal. 2020, 10, 4181–4192. [Google Scholar] [CrossRef]
- Mao, Z.; Campbell, C.T. The degree of rate control of catalyst-bound intermediates in catalytic reaction mechanisms: Relationships to site coverage. J. Catal. 2020, 381, 53–62. [Google Scholar] [CrossRef]
- Foley, B.L.; Bhan, A. Degree of rate control and De Donder relations—An interpretation based on transition state theory. J. Catal. 2020, 384, 231–251. [Google Scholar] [CrossRef]
- Vilekar, S.A.; Fishtik, I.; Datta, R. The steady-state kinetics of a catalytic reaction sequence. Chem. Eng. Sci. 2009, 64, 1968–1979. [Google Scholar] [CrossRef]
- Vilekar, S.A.; Fishtik, I.; Datta, R. The peculiar catalytic sequence of the ammonia decomposition reaction and its steady-state kinetics. Chem. Eng. Sci. 2012, 71, 333–344. [Google Scholar] [CrossRef]
- Vilekar, S.A.; Fishtik, I.; Datta, R. Topological analysis of catalytic reaction networks: Methanol decomposition on Pt (111). J. Catal. 2007, 252, 258–270. [Google Scholar] [CrossRef]
- Konsler, R.G.; Karl, J.; Jacobsen, E.N. Coperative asymmetric catalysis with dimeric salen complexes. J. Am. Chem. Soc. 1998, 120, 10780–10781. [Google Scholar] [CrossRef]
- Rosner, T.; le Bars, J.; Pfaltz, A.; Blackmond, D.G. Kinetic studies of Heck coupling reactions using palladacycle catalysts: Experimental and kinetic modelling of the role of dimer species. J. Am. Chem. Soc. 1998, 123, 1848–1855. [Google Scholar] [CrossRef]
- Noble-Teran, M.E.; Buhse, T.; Cruz, J.-M.; Coudret, C.; Micheau, J.-C. Nonlinear effects in asymmetric synthesis: A practical tool for the discrimination between monomer and dimer catalysis. ChemCatChem 2016, 8, 1836–1845. [Google Scholar] [CrossRef]
- Temkin, M. The kinetics of some industrial heterogeneous catalytic reactions. Adv. Catal. 1979, 28, 173–291. [Google Scholar]
- Horiuti, J.; Nakamura, T. On the theory of heterogeneous catalysis. Adv. Catal. 1967, 17, 1–74. [Google Scholar]
- Temkin, O.N. On Various Interconnections between Kinetics and Thermodynamics; LAP Lambert Academi Publishing: Saarbrueken, Germany, 2016; p. 119. [Google Scholar]
- Murzin, D.Y.; Touroude, R. On the rate of heterogeneous catalytic reactions with ionic intermediates. Catal. Lett. 1993, 20, 185–190. [Google Scholar] [CrossRef]
- Carucci, J.R.H.; Kurman, A.; Karhu, H.; Arve, K.; Eränen, K.; Wärnå, J.; Salmi, T.; Murzin, D.Y. Kinetics of the biofuels-assisted SCR of NOx over Ag/alumina-coated microchannels. Chem. Eng. J. 2009, 154, 34–44. [Google Scholar] [CrossRef]
- Murzin, D.Y. Interpretation of rate optima vs reaction parameters in steady state catalytic kinetics: Molecular aspects beyond concentration dependences. Mol. Catal. 2017, 433, 321–333. [Google Scholar] [CrossRef]
- Lazman, M.Z.; Yablonsky, G.S. Overall reaction rate equation of single-route complex catalytic reaction in terms of hypergeometric series. Adv. Chem. Eng. 2008, 34, 47–102. [Google Scholar]
- Laidler, K.J. Rate-controlling step: A necessary or useful concept? J. Chem. Educ. 1988, 65, 250–254. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Marin, G.B.; Yablonsky, G.S.; Constales, D. Kinetics of Chemical Reactions: Decoding Complexity, 2nd ed.; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Temkin, M.I.; Murzin, D.Y.; Kul’kova, N.V. Kinetics and mechanism of liquid-phase hydrogenation. Doklady Akademii Nauk SSSR 1988, 303, 659–662. [Google Scholar]
- Murzin, D.Y.; Sokolova, N.A.; Kul’kova, N.V.; Temkin, M.I. Kinetics of liquid-phase hydrogenation of benzene and toluene on a nickel catalyst. Kinet. Catal. 1989, 30, 1352. [Google Scholar]
- Temkin, M.I.; Murzin, D.Y.; Kul’kova, N.V. Mechanism of liquid-phase hydrogenation of a benzene ring. Kinet. Catal. 1989, 30, 637. [Google Scholar]
- Murzin, D.Y.; Kul’kova, N.V. Kinetics and mechanism of the liquid-phase hydrogenation. Kinet. Catal. 1995, 36, 70–76. [Google Scholar]
- Murzin, D.Y.; Kul’kova, N.V. Non-equilibrium effects in the liquid-phase catalytic hydrogenation. Catal. Today 1995, 24, 35. [Google Scholar] [CrossRef]
- Behravesh, E.; Melander, M.M.; Wärnå, J.; Salmi, T.; Honkala, K.; Murzin, D.Y. Oxidative dehydrogenation of ethanol on gold: Combination of kinetic experiments and computation approach to unravel the reaction mechanism. J. Catal. 2020. [Google Scholar] [CrossRef]
- Mulligan, C.J.; Parker, J.S.; Hii, K.K. Revisiting the mechanism of the Fujiwa-Moritani reaction. React. Chem. Eng. 2020, 5, 1104–1111. [Google Scholar] [CrossRef]
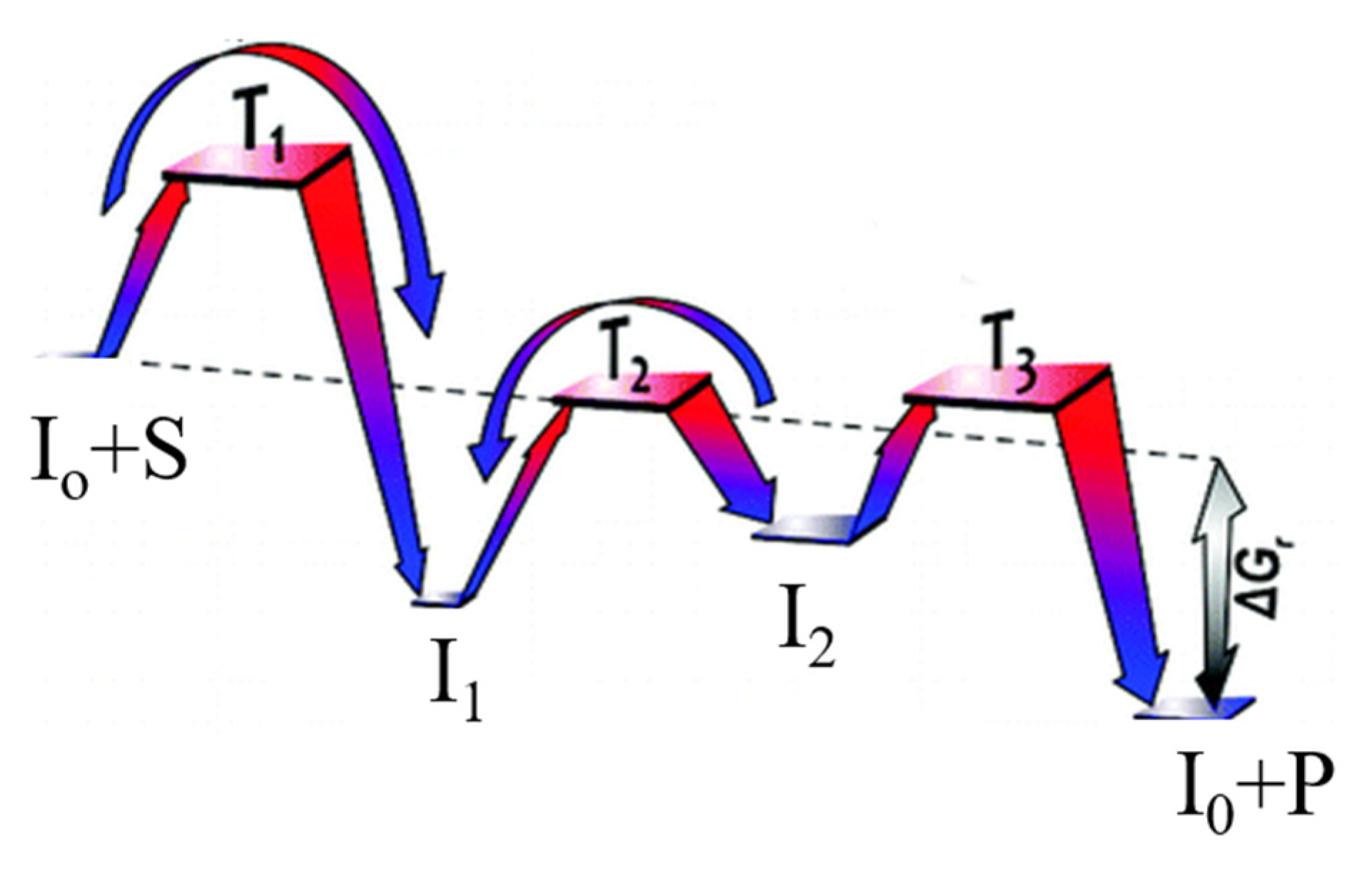
Figure 1.
Potential energy diagram using Gibbs energy and Gibbs activation energy for the reaction S->P. T1–T3 represent transition states, and I1–I2 intermediates, I0 is the free form of catalyst. Adapted from [40].
Figure 1.
Potential energy diagram using Gibbs energy and Gibbs activation energy for the reaction S->P. T1–T3 represent transition states, and I1–I2 intermediates, I0 is the free form of catalyst. Adapted from [40].

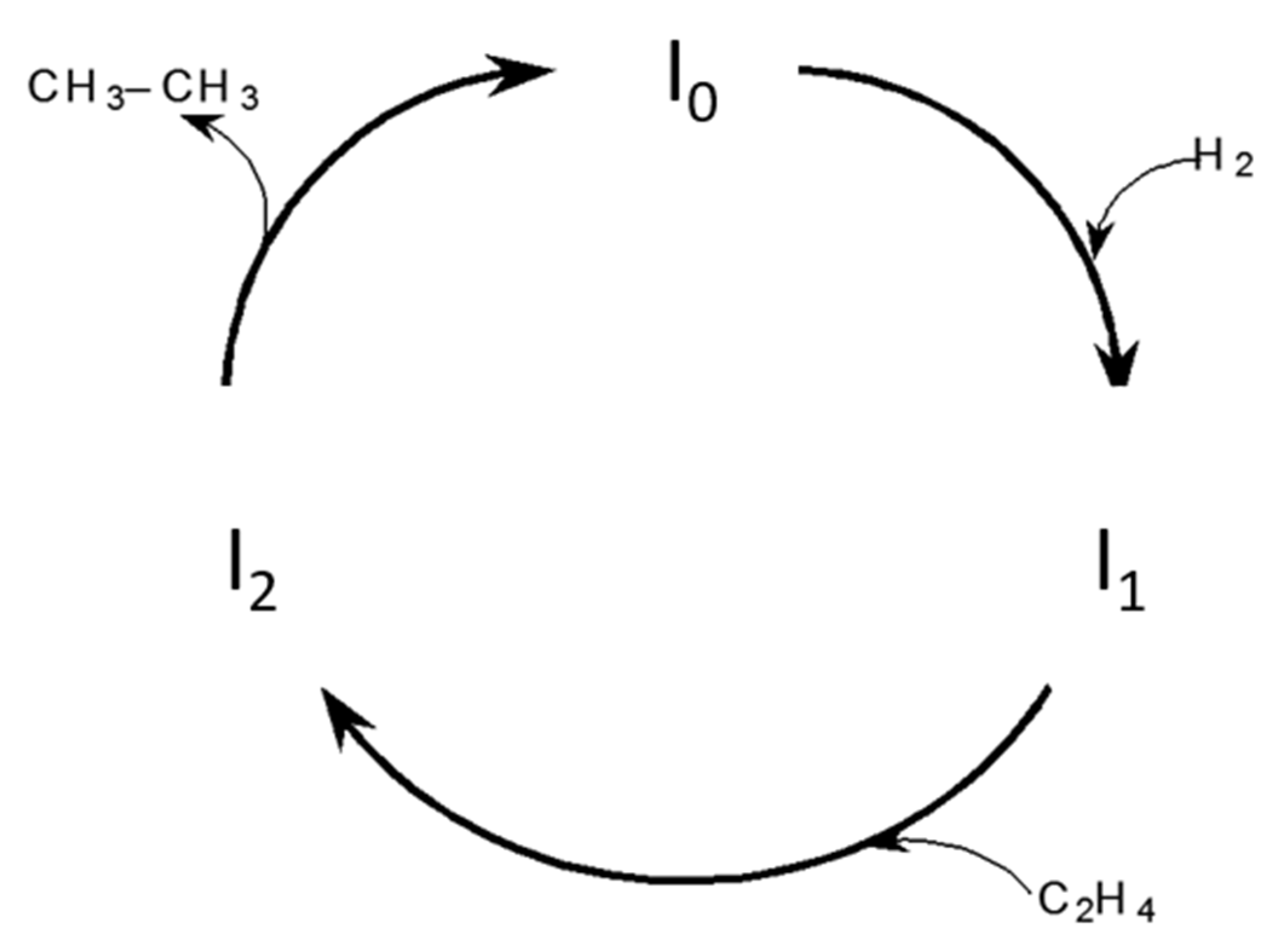
Figure 2.
Three-step direct ethylene hydrogenation mechanisms.

Figure 3.
The minimum energy path for ethanol (EtOH) dehydrogenation. The black and orange bars correspond to minima and transition states, respectively. * denotes adsorbed species, while (g) refers to gas phase species. Square brackets enclose a transition state structure where ‘ - -’ marks the bond being broken. Reproduced with permission from [47].
Figure 3.
The minimum energy path for ethanol (EtOH) dehydrogenation. The black and orange bars correspond to minima and transition states, respectively. * denotes adsorbed species, while (g) refers to gas phase species. Square brackets enclose a transition state structure where ‘ - -’ marks the bond being broken. Reproduced with permission from [47].

Figure 4.
Reaction mechanism for the Fujiwara–Moritani reaction [48]. Published by The Royal Society of Chemistry.
Figure 4.
Reaction mechanism for the Fujiwara–Moritani reaction [48]. Published by The Royal Society of Chemistry.

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Murzin, D.Y. Requiem for the Rate-Determining Step in Complex Heterogeneous Catalytic Reactions? Reactions 2020, 1, 37-46. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1010004
AMA Style
Murzin DY. Requiem for the Rate-Determining Step in Complex Heterogeneous Catalytic Reactions? Reactions. 2020; 1(1):37-46. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1010004
Chicago/Turabian StyleMurzin, Dmitry Yu. 2020. "Requiem for the Rate-Determining Step in Complex Heterogeneous Catalytic Reactions?" Reactions 1, no. 1: 37-46. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1010004