Ionic Liquids Based on Oxidoperoxido-Molybdenum(VI) Complexes with a Chelating Picolinate Ligand for Catalytic Epoxidation

 ,
,  , , and
, , and
Abstract
:
1. Introduction
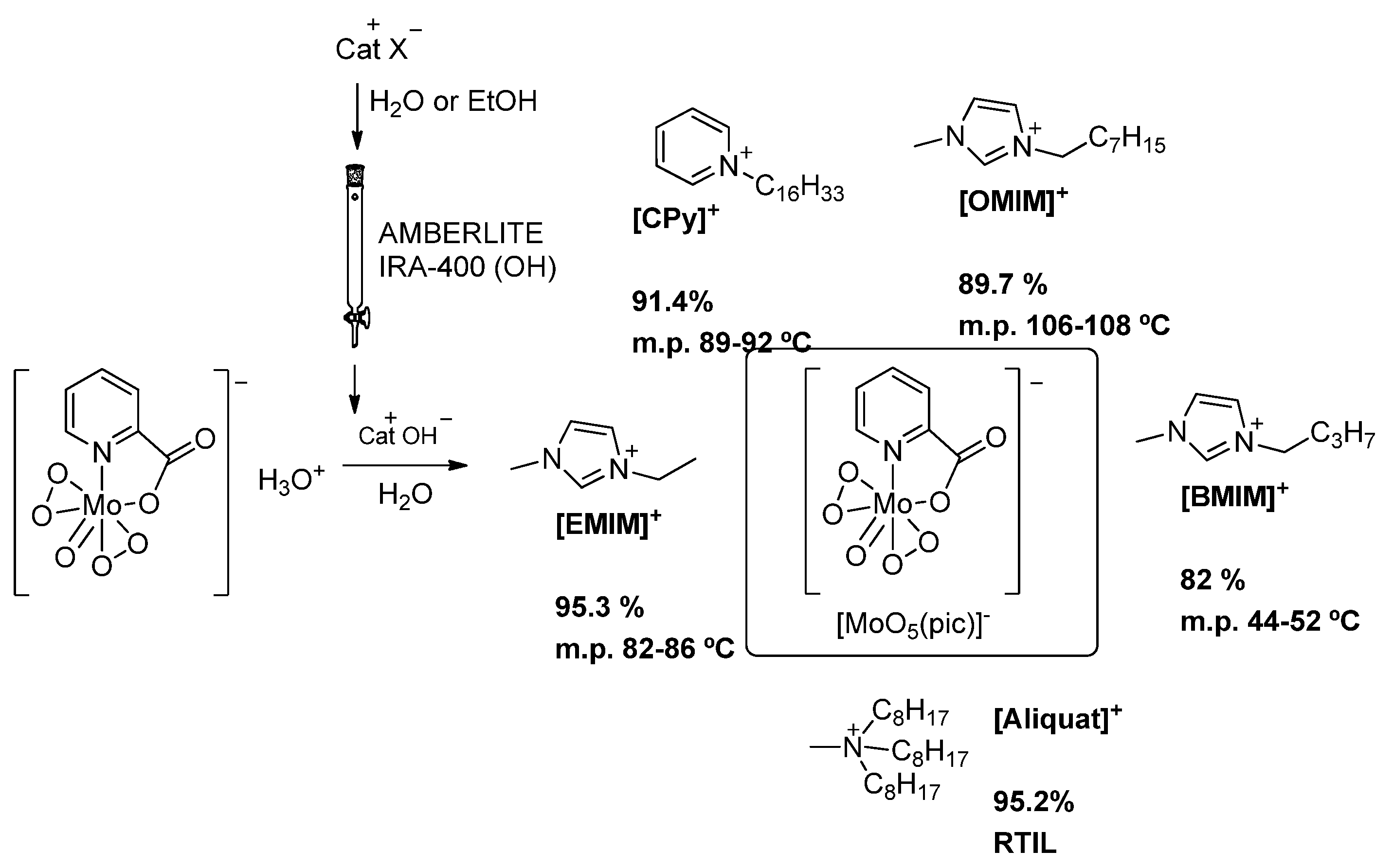
2. Materials and Methods
3. Results and Discussion
3.1. Characterization of the Ionic Complexes
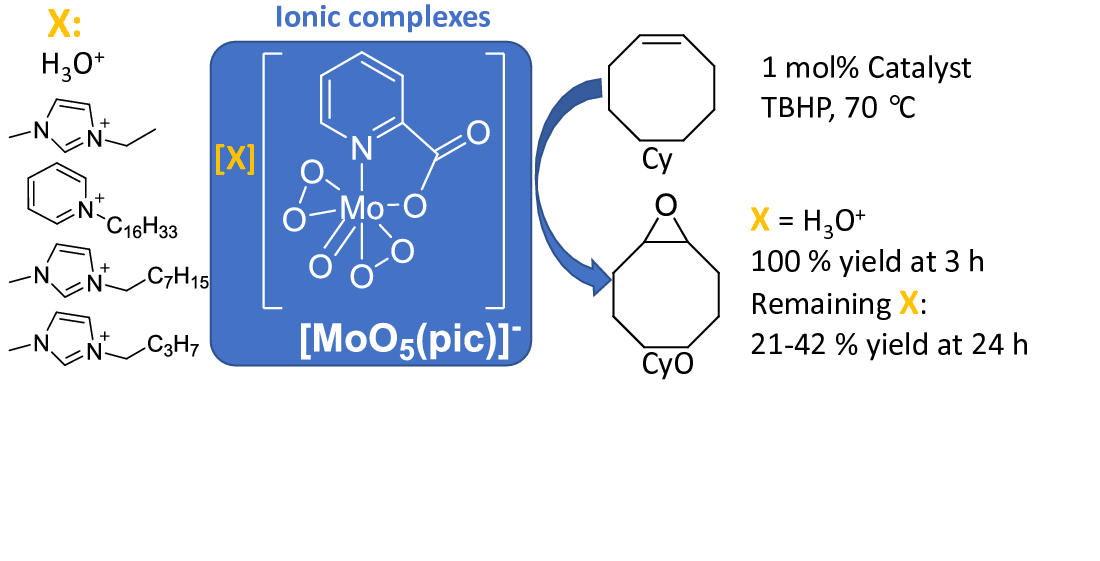
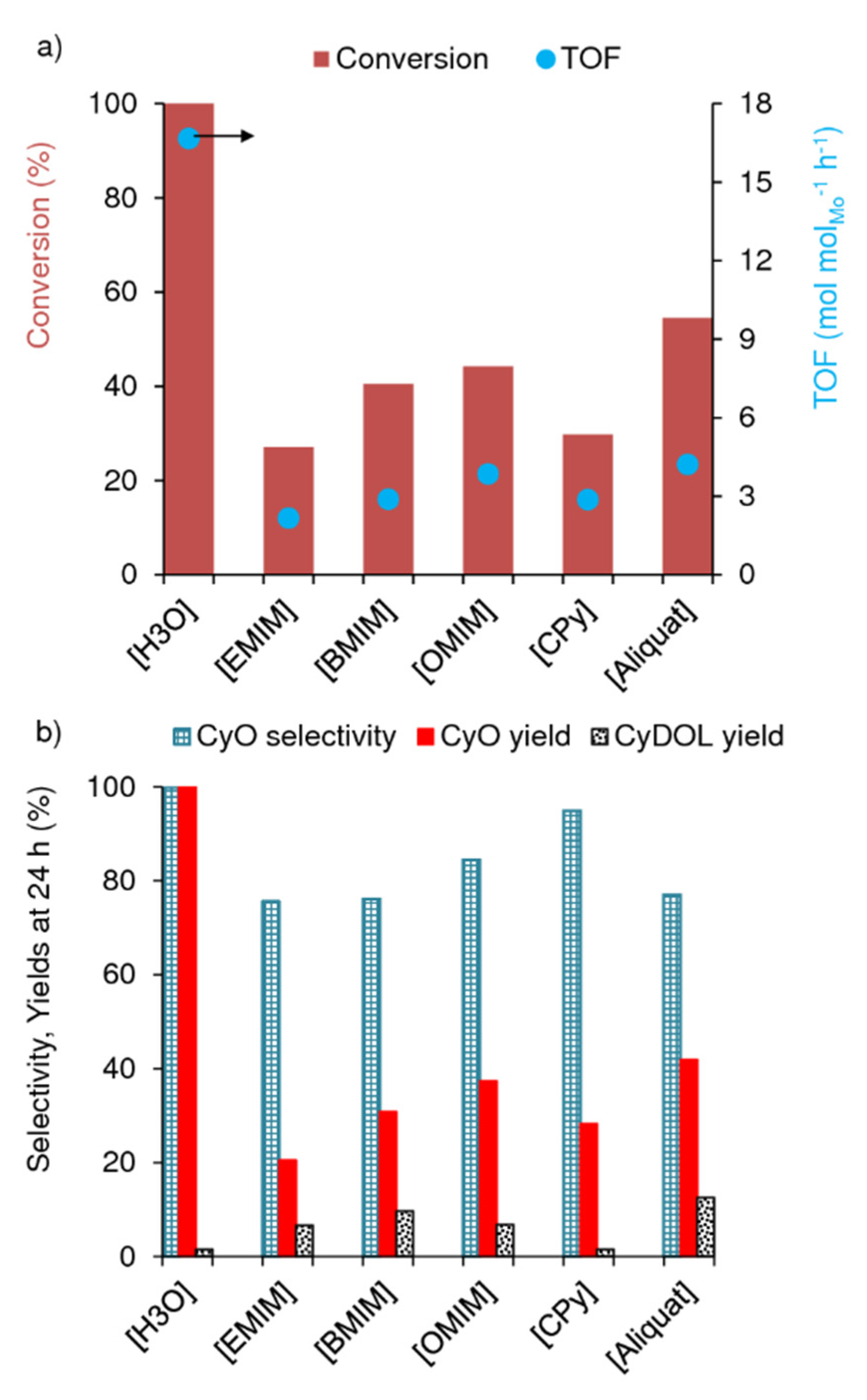
3.2. cis-Cyclooctene Catalytic Epoxidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catal [a] | Ox:Cy [b] | Mo [b] (mol%) | Solvent | T (°C) [c] | t (h) [c] | Conversion (%) [d] | Selectivity (%) [e] | Ref | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1.52 | 1 | TFT | 70 | 1/2/3 | 82/93/100 | 100/100/100 | This work |
| 2 | 2 | 1.52 | 1 | TFT | 70 | 6/24 | 13/27 | 94/76 | This work |
| 3 | 3 | 1.52 | 1 | TFT | 70 | 6/24 | 17/41 | 82/76 | This work |
| 4 | 4 | 1.52 | 1 | TFT | 70 | 6/24 | 23/44 | 77/85 | This work |
| 5 | 5 | 1.52 | 1 | TFT | 70 | 6/24 | 17/30 | 87/95 | This work |
| 6 | 6 | 1.52 | 1 | TFT | 70 | 6/24 | 25/55 | 84/77 | This work |
| 7 | 6 | 1.52 | 1 | MeCN | 70 | 6/24 | 5/27 | 100/76 | This work |
| 8 | 6 | 1.52 | 1 | DCE | 70 | 6/24 | 16/39 | 91/81 | This work |
| 9 | [MoO(O2)2(pbim)] | 1.53 | 1 | DCE | 70 | 6/24 | 81/98 | 100 | [72] |
| 10 | [MoO(O2)2(κ2-N,O-L)2] | 1.5 | 2.5 | Toluene | 25 | 22 | 15/38 | 100 | [73] |
| 11 | [MoO(O2)2(pzpyR1) | 1.0 | 0.4 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 12 | [MoO(O2)2(pzpyR2) | 1.0 | 0.4 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 13 | [MoO(O2)2(pzpyR3) | 1.0 | 0.4 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 14 | [MoO(O2)2(pzpyR4) | 1.0 | 0.3 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 15 | [MoO(O2)2(pzpyR5) | 1.0 | 0.2 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 16 | [MoO(O2)2(pzpyR6) | 1.0 | 0.4 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 17 | [MoO(O2)2(pzpyR7) | 1.0 | 0.2 | CHCl3 | 65 | 1 | TON [f] | 100 | [74] |
| 18 | [MoO(O2)2(pzpy)] | 1.52 | 0.3 | DCE | 75 | 6 | 100 | 100 | [39] |
| 19 | [MoO(O2)2(pypzEA)] | 1.52 | 0.1 | DCE | 55 | 24 | 22 | 100 | [42] |
| 20 | [MoO(O2)2(bzpypz)] | 1 | 0.5 | CHCl3 | nm [g] | 6 | 95 | 100 | [41] |
| 21 | [MoO(O2)2(pent-pp)] | 1.52 | 0.9 | DCE | 55 | 6 | 90 | 100 | [75] |
| 22 | [MoO(O2)2(2,2′-bipy)] | 1.5 | 1 | TFT | 55 | 6 | 34 | 100 | [76] |
| 23 | [MoO(O2)2(2,2′-bipy)] | 1.53 | 1 | H2O | 70 | 24 | 28 | 100 | [35] |
| 24 | [MoO(O2)2(di-tBu-bipy)] | 1.53 | 1 | H2O | 70 | 24 | 98 | 100 | [35] |
| 25 | [MoO(O2)2(BPM) | 1.53 | 1 | - | 55 | 6 | 100 | 100 | [45] |
| 26 | [nm][MoO(O2)2(phox)] [g] | 1 | 0.05 | - | 95 | 0.33 | 97 | 100 | [43] |
| 27 | [nm][MoO(O2)2(phox)] [g] | 1 | 0.05 | - | 80/85/95 | 0.33 | 45/69/99 | 100 | [44] |
| 28 | [nm][MoO(O2)2(phox)] [g] | 1 | 0.05 | DCE | 95 | 0.33 | 98 | 100 | [44] |
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mimoun, H.; Deroch, I.S.; Sajus, L. New molybdenum-6 and Tungsten-6 Peroxy-Complexes. Bull. Soc. Chim. Fr. 1969, 5, 1481–1492. [Google Scholar]
- Herbert, M.; Montilla, F.; Álvarez, E.; Galindo, A. New insights into the mechanism of oxodiperoxomolybdenum catalysed olefin epoxidation and the crystal structures of several oxo–peroxo molybdenum complexes. Dalton Trans. 2012, 41, 6942–6956. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.K.; Malik, K.M.A.; Gupta, S.; Chakraborty, S.; Ganguli, A.K.; Mukherjee, A.K.; Bhattacharyya, R. Oxo- and Oxoperoxo-molybdenum(VI) Complexes with Aryl Hydroxamates: Synthesis, Structure, and Catalytic Uses in Highly Efficient, Selective, and Ecologically Benign Peroxidic Epoxidation of Olefins. Inorg. Chem. 2006, 45, 9843–9857. [Google Scholar] [CrossRef] [PubMed]
- Salles, L.; Piquemal, J.-Y.; Thouvenot, R.; Minot, C.; Brégeault, J.-M. Catalytic epoxidation by heteropolyoxoperoxo complexes: From novel precursors or catalysts to a mechanistic approach. J. Mol. Catal. A Chem. 1997, 117, 375–387. [Google Scholar] [CrossRef]
- Joergensen, K.A. Transition-metal-catalyzed epoxidations. Chem. Rev. 1989, 89, 431–458. [Google Scholar] [CrossRef]
- Deubel, D.V.; Frenking, G.; Gisdakis, P.; Herrmann, W.A.; Rösch, N.; Sundermeyer, J. Olefin Epoxidation with Inorganic Peroxides. Solutions to Four Long-Standing Controversies on the Mechanism of Oxygen Transfer. Acc. Chem. Res. 2004, 37, 645–652. [Google Scholar] [CrossRef]
- Deubel, D.V. Ethylene Epoxidation with Tungsten Diperoxo Complexes: Is Relativity the Origin of Reactivity? J. Phys. Chem. A 2001, 105, 4765–4772. [Google Scholar] [CrossRef]
- Abednatanzi, S.; Abbasi, A.; Masteri-Farahani, M. Enhanced catalytic activity of nanoporous Cu3(BTC)2 metal-organic framework via immobilization of oxodiperoxo molybdenum complex. New J. Chem. 2015, 39, 5322–5328. [Google Scholar] [CrossRef]
- Das, S.; Bhowmick, T.; Punniyamurthy, T.; Dey, D.; Nath, J.; Chaudhuri, M.K. Molybdenum(VI)-peroxo complex catalyzed oxidation of alkylbenzenes with hydrogen peroxide. Tetrahedron Lett. 2003, 44, 4915–4917. [Google Scholar] [CrossRef]
- Bandyopadhyay, R.; Biswas, S.; Bhattacharyya, R.; Guha, S.; Mukherjee, A.K. Novel oxo-peroxo molybdenum(VI) complexes incorporating 8-quinolinol: Synthesis, structure and catalytic uses in the environmentally benign and cost-effective oxidation method of methyl benzenes: Ar(CH3) (n = 1, 2). Chem. Commun. 1999, 17, 1627–1628. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Barbuzzi, E.G.M.; Tomaselli, G.A.; Toscano, R.M. Multiplicity of Reaction Pathways in the Processes of Oxygen Transfer to Secondary Amines by Mo(VI) and W(VI) Peroxo Complexes. J. Org. Chem. 1996, 61, 6381–6387. [Google Scholar] [CrossRef]
- Biradar, A.V.; Kotbagi, T.V.; Dongare, M.K.; Umbarkar, S.B. Selective N-oxidation of aromatic amines to nitroso derivatives using a molybdenum acetylide oxo-peroxo complex as catalyst. Tetrahedron Lett. 2008, 49, 3616–3619. [Google Scholar] [CrossRef]
- Maiti, S.K.; Banerjee, S.; Mukherjee, A.K.; Abdul Malik, K.M.; Bhattacharyya, R. Oxoperoxo molybdenum(vi) and tungsten(vi) and oxodiperoxo molybdate(vi) and tungstate(vi) complexes with 8-quinolinol: Synthesis, structure and catalytic activity. New J. Chem. 2005, 29, 554–563. [Google Scholar] [CrossRef]
- Thiruvengetam, P.; Chakravarthy, R.D.; Chand, D.K. A molybdenum based metallomicellar catalyst for controlled and chemoselective oxidation of activated alcohols in aqueous medium. J. Catal. 2019, 376, 123–133. [Google Scholar] [CrossRef]
- Luan, Y.; Wang, G.; Luck, R.L.; Yang, M.; Han, X. Oxidation of Alcohols with Hydrogen Peroxide Catalyzed by Molybdenum(VI)–Peroxo Complex under Solvent-free Conditions. Chem. Lett. 2007, 36, 1236–1237. [Google Scholar] [CrossRef]
- Rostamnia, S.; Mohsenzad, F. Nanoarchitecturing of open metal site Cr-MOFs for oxodiperoxo molybdenum complexes [MoO(O2)2@En/MIL-100(Cr)] as promising and bifunctional catalyst for selective thioether oxidation. Mol. Catal. 2018, 445, 12–20. [Google Scholar] [CrossRef]
- Amini, M.; Bagherzadeh, M.; Atabaki, B.; Derakhshandeh, P.G.; Ellern, A.; Woo, L.K. Molybdenum(VI)–oxodiperoxo complex containing an oxazine ligand: Synthesis, X-ray studies, and catalytic activity. J. Coord. Chem. 2014, 67, 1429–1436. [Google Scholar] [CrossRef]
- Carrasco, C.J.; Montilla, F.; Bobadilla, L.; Ivanova, S.; Odriozola, J.A.; Galindo, A. Oxodiperoxomolybdenum complex immobilized onto ionic liquid modified SBA-15 as an effective catalysis for sulfide oxidation to sulfoxides using hydrogen peroxide. Catal. Today 2015, 255, 102–108. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Tomaselli, G.A.; Toscano, R.M.; Conte, V.; Di Furia, F. Application of the thianthrene 5-oxide mechanistic probe to peroxometal complexes. J. Am. Chem. Soc. 1991, 113, 6209–6212. [Google Scholar] [CrossRef]
- Bonchio, M.; Conte, V.; Assunta De Conciliis, M.; Di Furia, F.; Ballistreri, F.P.; Tomaselli, G.A.; Toscano, R.M. The Relative Reactivity of Thioethers and Sulfoxides toward Oxygen Transfer Reagents: The Oxidation of Thianthrene 5-Oxide and Related Compounds by MoO5HMPT. J. Org. Chem. 1995, 60, 4475–4480. [Google Scholar] [CrossRef]
- Basak, A.; Barlan, A.U.; Yamamoto, H. Catalytic enantioselective oxidation of sulfides and disulfides by a chiral complex of bis-hydroxamic acid and molybdenum. Tetrahedron Asymmetry 2006, 17, 508–511. [Google Scholar] [CrossRef]
- Batigalhia, F.; Zaldini-Hernandes, M.; Ferreira, A.; Malvestiti, I.; Cass, Q. Selective and Mild Oxidation of Sulfides to Sulfoxides by Oxodiperoxo Molybdenum Complexes Adsorbed onto Silica Gel. Tetrahedron 2001, 57, 9669–9676. [Google Scholar] [CrossRef]
- Bortolini, O.; Di Furia, F.; Modena, G.; Seraglia, R. Metal catalysis in oxidation by peroxides. Sulfide oxidation and olefin epoxidation by dilute hydrogen peroxide, catalyzed by molybdenum and tungsten derivatives under phase-transfer conditions. J. Org. Chem. 1985, 50, 2688–2690. [Google Scholar] [CrossRef]
- Keilen, G.; Benneche, T.; Gaare, K.; Undheim, K. Peroxymolybdenum complexes in sulfide to sulfone oxidations. Acta Chem. Scand. 1992, 46, 867–871. [Google Scholar] [CrossRef]
- Khurana, J.M.; Agrawal, A.; Kumar, S. Oxidation of chalcogenides using the peroxo complex of molybdenum [MoO(O2)2 (H2O) (hmpa)], hmpa = hexamethylphosphoramide. J. Braz. Chem. Soc. 2009, 20, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- Burke, A.J. Chiral oxoperoxomolybdenum(VI) complexes for enantioselective olefin epoxidation: Some mechanistic and stereochemical reflections. Coord. Chem. Rev. 2008, 252, 170–175. [Google Scholar] [CrossRef]
- Amini, M.; Haghdoost, M.M.; Bagherzadeh, M. Monomeric and dimeric oxido–peroxido tungsten(VI) complexes in catalytic and stoichiometric epoxidation. Coord. Chem. Rev. 2014, 268, 83–100. [Google Scholar] [CrossRef]
- Amini, M.; Haghdoost, M.M.; Bagherzadeh, M. Oxido-peroxido molybdenum(VI) complexes in catalytic and stoichiometric oxidations. Coord. Chem. Rev. 2013, 257, 1093–1121. [Google Scholar] [CrossRef]
- Dickman, M.H.; Pope, M.T. Peroxo and Superoxo Complexes of Chromium, Molybdenum, and Tungsten. Chem. Rev. 1994, 94, 569–584. [Google Scholar] [CrossRef]
- Neves, P.; Gomes, A.C.; Paz, F.A.A.; Valente, A.A.; Gonçalves, I.S.; Pillinger, M. Synthesis, structure and catalytic olefin epoxidation activity of a dinuclear oxo-bridged oxodiperoxomolybdenum(VI) complex containing coordinated 4,4′-bipyridinium. Mol. Catal. 2017, 432, 104–114. [Google Scholar] [CrossRef]
- Gharah, N.; Chattopadhyay, B.; Maiti, S.K.; Mukherjee, M. Synthesis and catalytic epoxidation potential of oxodiperoxo molybdenum(VI) complexes with 2-hydroxybenzohydroxamate and 2-hydroxybenzoate: The crystal structure of PPh4[MoO(O2)2 (HBA)]. Transit. Metal Chem. 2010, 35, 531–539. [Google Scholar] [CrossRef]
- Gharah, N.; Drew, M.G.B.; Bhattacharyya, R. Synthesis and catalytic epoxidation potentiality of oxodiperoxo molybdenum(VI) complexes with pyridine-2-carboxaldoxime and pyridine-2-carboxylate: The crystal structure of PMePh3[MoO(O2)2(PyCO)]. Transit. Metal Chem. 2009, 34, 549–557. [Google Scholar] [CrossRef]
- Maurya, M.R.; Kumar, M.; Sikarwar, S. Polymer-anchored oxoperoxo complexes of vanadium(V), molybdenum(VI) and tungsten(VI) as catalyst for the oxidation of phenol and styrene using hydrogen peroxide as oxidant. React. Funct. Polym. 2006, 66, 808–818. [Google Scholar] [CrossRef]
- Gharah, N.; Chakraborty, S.; Mukherjee, A.K.; Bhattacharyya, R. Highly efficient epoxidation method of olefins with hydrogen peroxide as terminal oxidant, bicarbonate as a co-catalyst and oxodiperoxo molybdenum(vi) complex as catalyst. Chem. Commun. 2004, 22, 2630–2632. [Google Scholar] [CrossRef]
- Gamelas, C.A.; Gomes, A.C.; Bruno, S.M.; Paz, F.A.A.; Valente, A.A.; Pillinger, M.; Romão, C.C.; Gonçalves, I.S. Molybdenum(vi) catalysts obtained from η3-allyl dicarbonyl precursors: Synthesis, characterization and catalytic performance in cyclooctene epoxidation. Dalton Trans. 2012, 41, 3474–3484. [Google Scholar] [CrossRef]
- Herbert, M.; Álvarez, E.; Cole-Hamilton, D.J.; Montilla, F.; Galindo, A. Olefin epoxidation by hydrogen peroxide catalysed by molybdenum complexes in ionic liquids and structural characterisation of the proposed intermediate dioxoperoxomolybdenum species. Chem. Commun. 2010, 46, 5933–5935. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.-F.; Wang, L.-S.; Fan, C.-L. Catalytic Epoxidation of a Technical Mixture of Methyl Oleate and Methyl Linoleate in Ionic Liquids Using MoO(O2)2•2QOH (QOH = 8-quinilinol) as Catalyst and NaHCO3 as co-Catalyst. Molecules 2009, 14, 2935–2946. [Google Scholar] [CrossRef]
- Herbert, M.; Montilla, F.; Galindo, A. Olefin epoxidation in solventless conditions and apolar media catalysed by specialised oxodiperoxomolybdenum complexes. J. Mol. Catal. A Chem. 2011, 338, 111–120. [Google Scholar] [CrossRef]
- Amarante, T.R.; Neves, P.; Gomes, A.C.; Nolasco, M.M.; Ribeiro-Claro, P.; Coelho, A.C.; Valente, A.A.; Paz, F.A.A.; Smeets, S.; McCusker, L.B.; et al. Synthesis, Structural Elucidation, and Catalytic Properties in Olefin Epoxidation of the Polymeric Hybrid Material [Mo3O9(2-[3(5)-Pyrazolyl]pyridine)]n. Inorg. Chem. 2014, 53, 2652–2665. [Google Scholar] [CrossRef]
- da Palma Carreiro, E.; Yong-En, G.; Burke, A.J. Synthesis, characterisation and reactivity of oxodiperoxo-[2-(1-pyrazolyl)-6-menthylpyridine]molybdenum(VI): The first chiral 2-(1-pyrazole)pyridineoxodiperoxomolybdenum(VI) complex. Inorg. Chim. Acta 2006, 359, 1519–1523. [Google Scholar] [CrossRef]
- Hinner, M.J.; Grosche, M.; Herdtweck, E.; Thiel, W.R. A Merrifield Resin Functionalized with Molybdenum Peroxo Complexes: Synthesis and Catalytic Properties. Z. Anorg. Allg. Chem. 2003, 629, 2251–2257. [Google Scholar] [CrossRef]
- Amarante, T.R.; Gomes, A.C.; Neves, P.; Paz, F.A.A.; Valente, A.A.; Pillinger, M.; Gonçalves, I.S. A dinuclear oxo-bridged molybdenum(VI) complex containing a bidentate pyrazolylpyridine ligand: Structure, characterization and catalytic performance for olefin epoxidation. Inorg. Chem. Commun. 2013, 32, 59–63. [Google Scholar] [CrossRef]
- Zare, M.; Moradi-Shoeili, Z.; Esmailpour, P.; Akbayrak, S.; Özkar, S. Oxazine containing molybdenum(VI)–oxodiperoxo complex immobilized on SBA-15 as highly active and selective catalyst in the oxidation of alkenes to epoxides under solvent-free conditions. Microporous Mesoporous Mater. 2017, 251, 173–180. [Google Scholar] [CrossRef]
- Zare, M.; Moradi-Shoeili, Z. Oxidation of alkenes catalysed by molybdenum(VI)–oxodiperoxo complex anchored on the surface of magnetic nanoparticles under solvent-free conditions. Appl. Organomet. Chem. 2017, 31, e3611. [Google Scholar] [CrossRef]
- Figueiredo, S.; Gomes, A.C.; Fernandes, J.A.; Paz, F.A.A.; Lopes, A.D.; Lourenço, J.P.; Pillinger, M.; Gonçalves, I.S. Bis(pyrazolyl)methanetetracarbonyl-molybdenum(0) as precursor to a molybdenum(VI) catalyst for olefin epoxidation. J. Organomet. Chem. 2013, 723, 56–64. [Google Scholar] [CrossRef]
- Herbert, M.; Montilla, F.; Galindo, A.; Moyano, R.; Pastor, A.; Álvarez, E. Influence of N-donor bases and the solvent in oxodiperoxomolybdenum catalysed olefin epoxidation with hydrogen peroxide in ionic liquids. Dalton Trans. 2011, 40, 5210–5219. [Google Scholar] [CrossRef]
- Shylesh, S.; Schweizer, J.; Demeshko, S.; Schünemann, V.; Ernst, S.; Thiel, W. Nanoparticle Supported, Magnetically Recoverable Oxodiperoxo Molybdenum Complexes: Efficient Catalysts for Selective Epoxidation Reactions. Adv. Synth. Catal. 2009, 351, 1789–1795. [Google Scholar] [CrossRef]
- Jia, M.; Seifert, A.; Berger, M.; Giegengack, H.; Schulze, S.; Thiel, W.R. Hybrid Mesoporous Materials with a Uniform Ligand Distribution: Synthesis, Characterization, and Application in Epoxidation Catalysis. Chem. Mater. 2004, 16, 877–882. [Google Scholar] [CrossRef]
- Jia, M.; Seifert, A.; Thiel, W.R. Sol–gel synthesis of oxodiperoxo molybdenum-modified organic–inorganic materials for the catalytic epoxidation of cyclooctene. J. Catal. 2004, 221, 319–324. [Google Scholar] [CrossRef]
- Jia, M.; Seifert, A.; Thiel, W.R. Mesoporous MCM-41 Materials Modified with Oxodiperoxo Molybdenum Complexes: Efficient Catalysts for the Epoxidation of Cyclooctene. Chem. Mater. 2003, 15, 2174–2180. [Google Scholar] [CrossRef]
- Jia, M.; Thiel, W.R. Oxodiperoxo molybdenum modified mesoporous MCM-41 materials for the catalytic epoxidation of cyclooctene. Chem. Commun. 2002, 20, 2392–2393. [Google Scholar] [CrossRef] [PubMed]
- Dengel, A.C.; Griffith, W.P.; Powell, R.D.; Skapski, A.C. Studies on transition-metal peroxo complexes. Part 7. Molybdenum(VI) and tungsten(VI) carboxylato peroxo complexes, and the X-ray crystal structure of K2[MoO(O2)2(glyc)]2H2O. J. Chem. Soc. Dalton Trans. 1987, 5, 991–995. [Google Scholar] [CrossRef]
- Bonchio, M.; Conte, V.; Furia, F.D.; Carofiglio, T.; Magno, F.; Pastore, P. CoII-induced radical oxidations by peroxomolybdenum complexes. J. Chem. Soc. Perkin Trans. 2 1993, 10, 1923–1926. [Google Scholar] [CrossRef]
- Campestrini, S.; Di Furia, F.; Novello, F. Oxidation of meso- and d,l-hydrobenzoin by peroxomolybdenum complexes: A mechanistic investigation. J. Mol. Catal. 1993, 78, 159–168. [Google Scholar] [CrossRef]
- Campestrini, S.; Di Furia, F.; Modena, G.; Bortolini, O. Metal catalysis in oxidation by peroxides. Part 33. Chemoselective alcohol oxidations by the anionic molybdenum-picolinate N-oxido peroxo complex MoO5PICO. J. Org. Chem. 1990, 55, 3658–3660. [Google Scholar] [CrossRef]
- Bortolini, O.; Campestrini, S.; Di Furia, F.; Modena, G.; Valle, G. Metal catalysis in oxidation by peroxides. 27. Anionic molybdenum-picolinate N-oxido-peroxo complex: An effective oxidant of primary and secondary alcohols in nonpolar solvents. J. Org. Chem. 1987, 52, 5467–5469. [Google Scholar] [CrossRef]
- Jacobson, S.E.; Muccigrosso, D.A.; Mares, F. Oxidation of alcohols by molybdenum and tungsten peroxo complexes. J. Org. Chem. 1979, 44, 921–924. [Google Scholar] [CrossRef]
- Di Furia, F.; Fornasier, R.; Tonellato, U. Catalytic oxidation by a Mo(VI)diperoxo complex as a counter-ion of cationic surfactants in dilute H2O2 aqueous solutions. J. Mol. Catal. 1983, 19, 81–84. [Google Scholar] [CrossRef]
- Campestrini, S.; Conte, V.; Di Furia, F.; Modena, G.; Bortolini, O. Metal catalysis in oxidation by peroxides. 30. Electrophilic oxygen transfer from anionic, coordinatively saturated molybdenum peroxo complexes. J. Org. Chem. 1988, 53, 5721–5724. [Google Scholar] [CrossRef]
- Gharah, N.; Chakraborty, S.; Mukherjee, A.K.; Bhattacharyya, R. Oxoperoxo molybdenum(VI)- and tungsten(VI) complexes with 1-(2′-hydroxyphenyl) ethanone oxime: Synthesis, structure and catalytic uses in the oxidation of olefins, alcohols, sulfides and amines using H2O2 as a terminal oxidant. Inorg. Chim. Acta 2009, 362, 1089–1100. [Google Scholar] [CrossRef]
- Jacobson, S.E.; Tang, R.; Mares, F. Group 6 transition metal peroxo complexes stabilized by polydentate pyridinecarboxylate ligands. Inorg. Chem. 1978, 17, 3055–3063. [Google Scholar] [CrossRef]
- Ramos, M.L.; Justino, L.L.G.; Burrows, H.D. Structural considerations and reactivity of peroxocomplexes of V(v), Mo(vi) and W(vi). Dalton Trans. 2011, 40, 4374–4383. [Google Scholar] [CrossRef] [PubMed]
- Galiński, M.; Lewandowski, A.; Stępniak, I. Ionic liquids as electrolytes. Electrochim. Acta 2006, 51, 5567–5580. [Google Scholar] [CrossRef]
- Matsuyama, S.; Kinugasa, S.; Tanabe, K.; Tamura, T. IR Spectroscopy, Spectral Database for Organic Compounds (SDBS); AIST: Tsukuba, Japan, 2020; Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (accessed on 1 December 2020).
- Veiros, L.F.; Prazeres, Â.; Costa, P.J.; Romão, C.C.; Kühn, F.E.; José Calhorda, M. Olefin epoxidation with tert-butyl hydroperoxide catalyzed by MoO2X2L complexes: A DFT mechanistic study. Dalton Trans. 2006, 11, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.O.; Sharpless, K.B. Mechanism of the molybdenum and vanadium catalyzed epoxidation of olefins by alkyl hydroperoxides. J. Org. Chem. 1977, 42, 1587–1590. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert-Butylhydroperoxide Activation. ChemCatChem 2013, 5, 601–611. [Google Scholar] [CrossRef]
- Calhorda, M.; Costa, P. Unveiling the Mechanisms of Catalytic Oxidation Reactions Mediated by Oxo-Molybdenum Complexes: A Computational Overview. Curr. Org. Chem. 2012, 16, 65–72. [Google Scholar] [CrossRef]
- Thiel, W.R. Metal catalyzed oxidations. Part 5. Catalytic olefin epoxidation with seven-coordinate oxobisperoxo molybdenum complexes: A mechanistic study. J. Mol. Catal. A Chem. 1997, 117, 449–454. [Google Scholar] [CrossRef]
- Thiel, W.R.; Eppinger, J. Molybdenum-Catalyzed Olefin Epoxidation: Ligand Effects. Chem. A Eur. J. 1997, 3, 696–705. [Google Scholar] [CrossRef]
- Thiel, W.R.; Priermeier, T. The First Olefin-Substituted Peroxomolybdenum Complex: Insight into a New Mechanism for the Molybdenum-Catalyzed Epoxidation of Olefins. Angew. Chem. Int. Ed. Engl. 1995, 34, 1737–1738. [Google Scholar] [CrossRef]
- Neves, P.; Nogueira, L.S.; Gomes, A.C.; Oliveira, T.S.M.; Lopes, A.D.; Valente, A.A.; Gonçalves, I.S.; Pillinger, M. Chemistry and Catalytic Performance of Pyridyl-Benzimidazole Oxidomolybdenum(VI) Compounds in (Bio)Olefin Epoxidation. Eur. J. Inorg. Chem. 2017, 2017, 2617–2627. [Google Scholar] [CrossRef]
- Brito, J.A.; Gómez, M.; Muller, G.; Teruel, H.; Clinet, J.-C.; Duñach, E.; Maestro, M.A. Structural Studies of Mono- and Dimetallic MoVI Complexes—A New Mechanistic Contribution in Catalytic Olefin Epoxidation Provided by Oxazoline Ligands. Eur. J. Inorg. Chem. 2004, 21, 4278–4285. [Google Scholar] [CrossRef]
- Thiel, W.R.; Angstl, M.; Priermeier, T. Substituierte N,N-Chelat-Liganden—Anwendungen in der Molybdän-katalysierten Olefin-Epoxidation. Chem. Ber. 1994, 127, 2373–2379. [Google Scholar] [CrossRef]
- Amarante, T.R.; Neves, P.; Paz, F.A.A.; Valente, A.A.; Pillinger, M.; Gonçalves, I.S. Investigation of a dichlorodioxomolybdenum(vi)-pyrazolylpyridine complex and a hybrid derivative as catalysts in olefin epoxidation. Dalton Trans. 2014, 43, 6059–6069. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.M.; Nogueira, L.S.; Gomes, A.C.; Valente, A.A.; Gonçalves, I.S.; Pillinger, M. High-yield synthesis and catalytic response of chainlike hybrid materials of the [(MoO3)m(2,2′-bipyridine)n] family. New J. Chem. 2018, 42, 16483–16492. [Google Scholar] [CrossRef] [Green Version]
- Dinoi, C.; Ciclosi, M.; Manoury, E.; Maron, L.; Perrin, L.; Poli, R. Olefin epoxidation by H2O2/MeCN catalysed by cyclopentadienyloxidotungsten(VI) and molybdenum(VI) complexes: Experiments and computations. Chem. Eur. J. 2010, 16, 9572–9584. [Google Scholar] [CrossRef]
- Sözen-Aktaş, P.; Manoury, E.; Demirhan, F.; Poli, R. Molybdenum versus Tungsten for the Epoxidation of Cyclooctene Catalyzed by [Cp*2M2O5]. Eur. J. Inorg. Chem. 2013, 15, 2728–2735. [Google Scholar] [CrossRef]
- Di Valentin, C.; Gisdakis, P.; Yudanov, I.V.; Rösch, N. Olefin Epoxidation by Peroxo Complexes of Cr, Mo, and W. A Comparative Density Functional Study. J. Org. Chem. 2000, 65, 2996–3004. [Google Scholar] [CrossRef]
- Amarante, T.R.; Antunes, M.M.; Valente, A.A.; Paz, F.A.A.; Pillinger, M.; Gonçalves, I.S. Crystal Structure and Catalytic Behavior in Olefin Epoxidation of a One-Dimensional Tungsten Oxide/Bipyridine Hybrid. Inorg. Chem. 2015, 54, 9690–9703. [Google Scholar] [CrossRef]




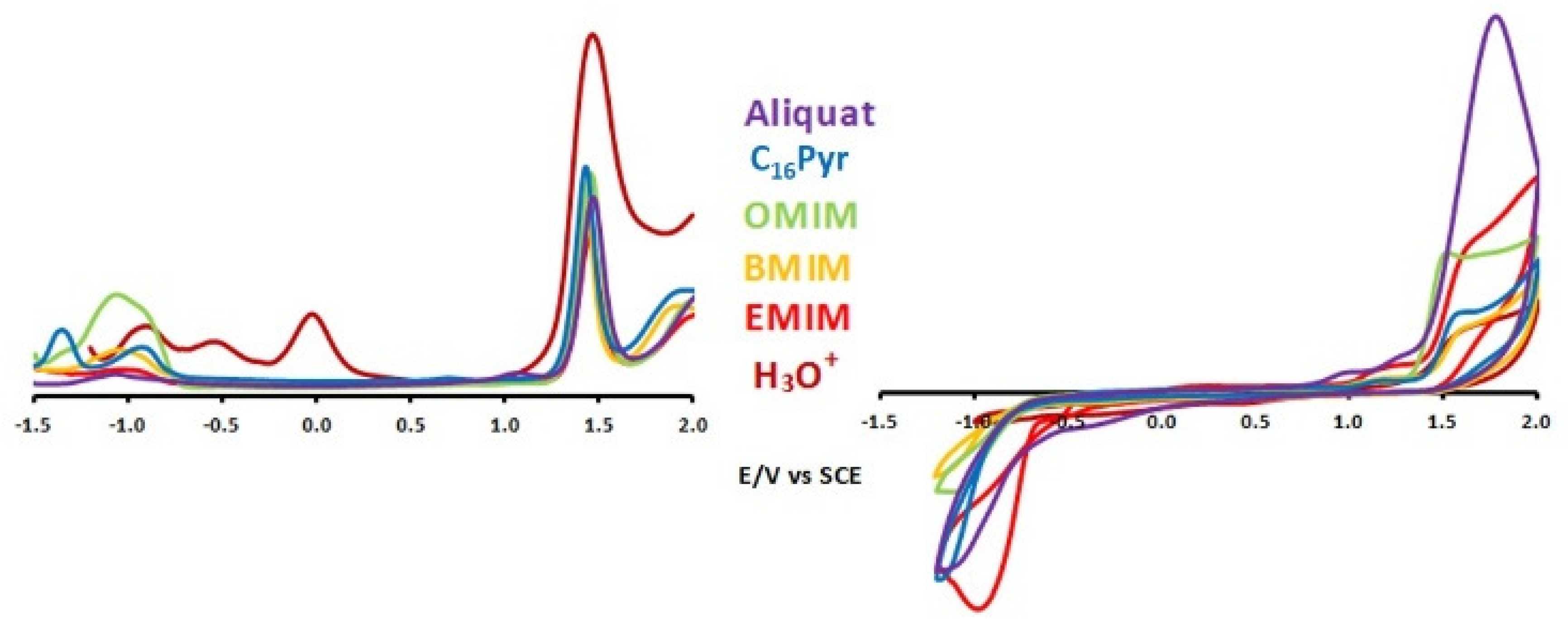
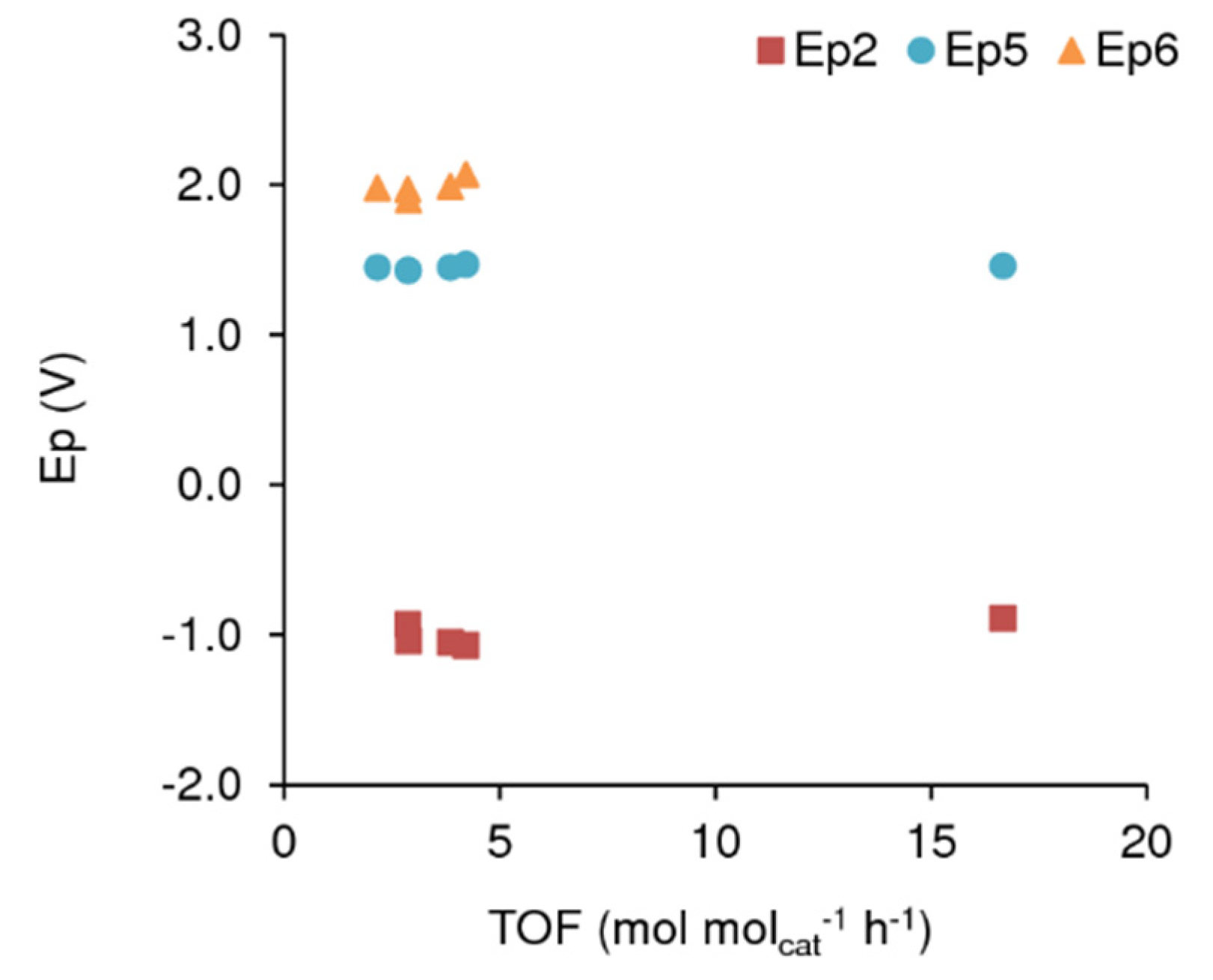
| [Cat]+ | Ep1 | Ep2 | Ep3 | Ep4 | Ep5 | Ep6 |
|---|---|---|---|---|---|---|
| [Aliquat]+ | −1.07 | +1.47 | +2.07 | |||
| [C16Py]+ | −1.36 | −0.93 | +1.43 | +1.97 | ||
| [OMIM]+ | −1.05 | +1.45 | +1.99 | |||
| [BMIM]+ | −1.04 | +1.43 | +1.90 | |||
| [EMIM]+ | +1.45 | +1.98 | ||||
| [H3O]+ | −0.89 | −0.55 | −0.02 | +1.46 | n.d. 1 |
| Substrate | Time (h) | Conv. [b] (%) | Select. [c] (%) |
|---|---|---|---|
| cis-Cyclooctene | 1/2/3 | 82/93/100 | 100/100/100 |
| trans-2-Octene | 1/3/4 | 35/78/84 | 100/97/95 |
| Cyclododecene | 1/3/4 | 33/65/71 | 97/95/95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrovski, Ž.; Antunes, M.M.; Mendo, A.S.; Cabrita, L.; Gonçalves, I.S.; Valente, A.A.; Branco, L.C. Ionic Liquids Based on Oxidoperoxido-Molybdenum(VI) Complexes with a Chelating Picolinate Ligand for Catalytic Epoxidation. Reactions 2020, 1, 147-161. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020012
Petrovski Ž, Antunes MM, Mendo AS, Cabrita L, Gonçalves IS, Valente AA, Branco LC. Ionic Liquids Based on Oxidoperoxido-Molybdenum(VI) Complexes with a Chelating Picolinate Ligand for Catalytic Epoxidation. Reactions. 2020; 1(2):147-161. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020012
Chicago/Turabian StylePetrovski, Željko, Margarida M. Antunes, Ana Soraia Mendo, Luís Cabrita, Isabel S. Gonçalves, Anabela A. Valente, and Luís C. Branco. 2020. "Ionic Liquids Based on Oxidoperoxido-Molybdenum(VI) Complexes with a Chelating Picolinate Ligand for Catalytic Epoxidation" Reactions 1, no. 2: 147-161. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020012