
Synthesis and Biological Activity Evaluation of Novel 5-Methyl-7-phenyl-3H-thiazolo[4,5-b]pyridin-2-ones


 ,
,  , , and
, , and 
Abstract
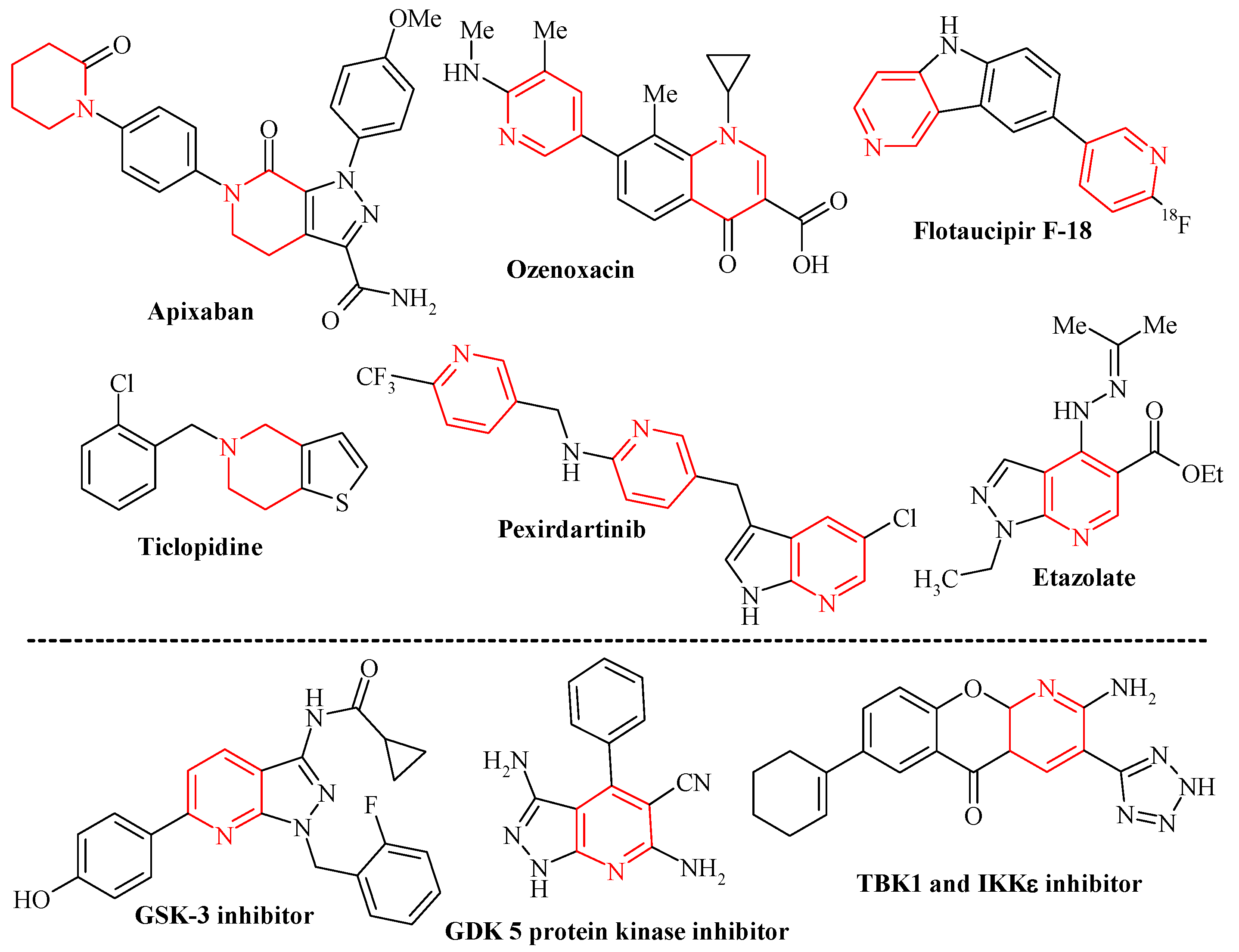
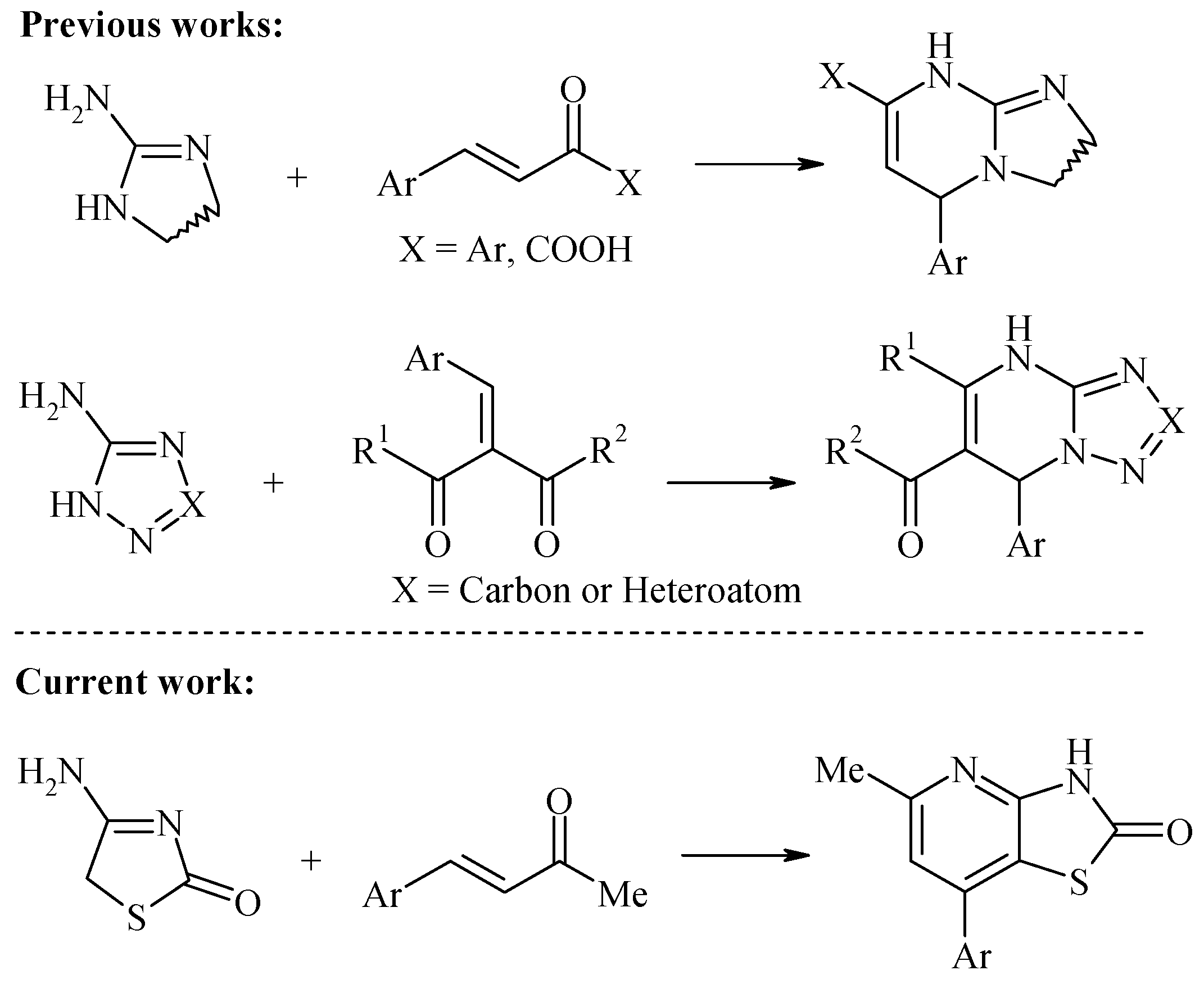
:1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Synthesis and Characterization of Compounds
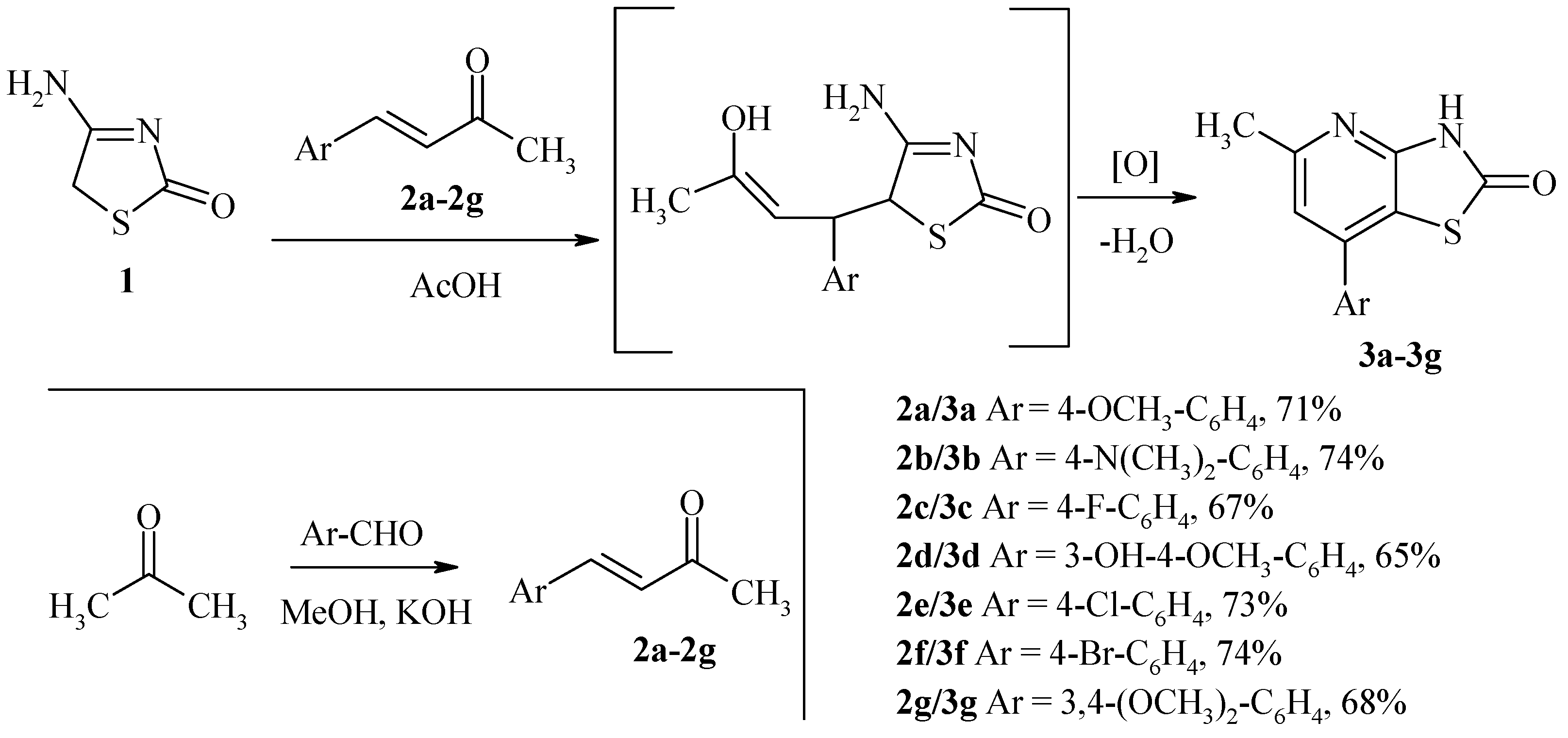
General Procedure for the Synthesis of 5-Methyl-7-Phenyl-3H-Thiazolo[4,5-b]Pyridin-2-Ones 3a–g
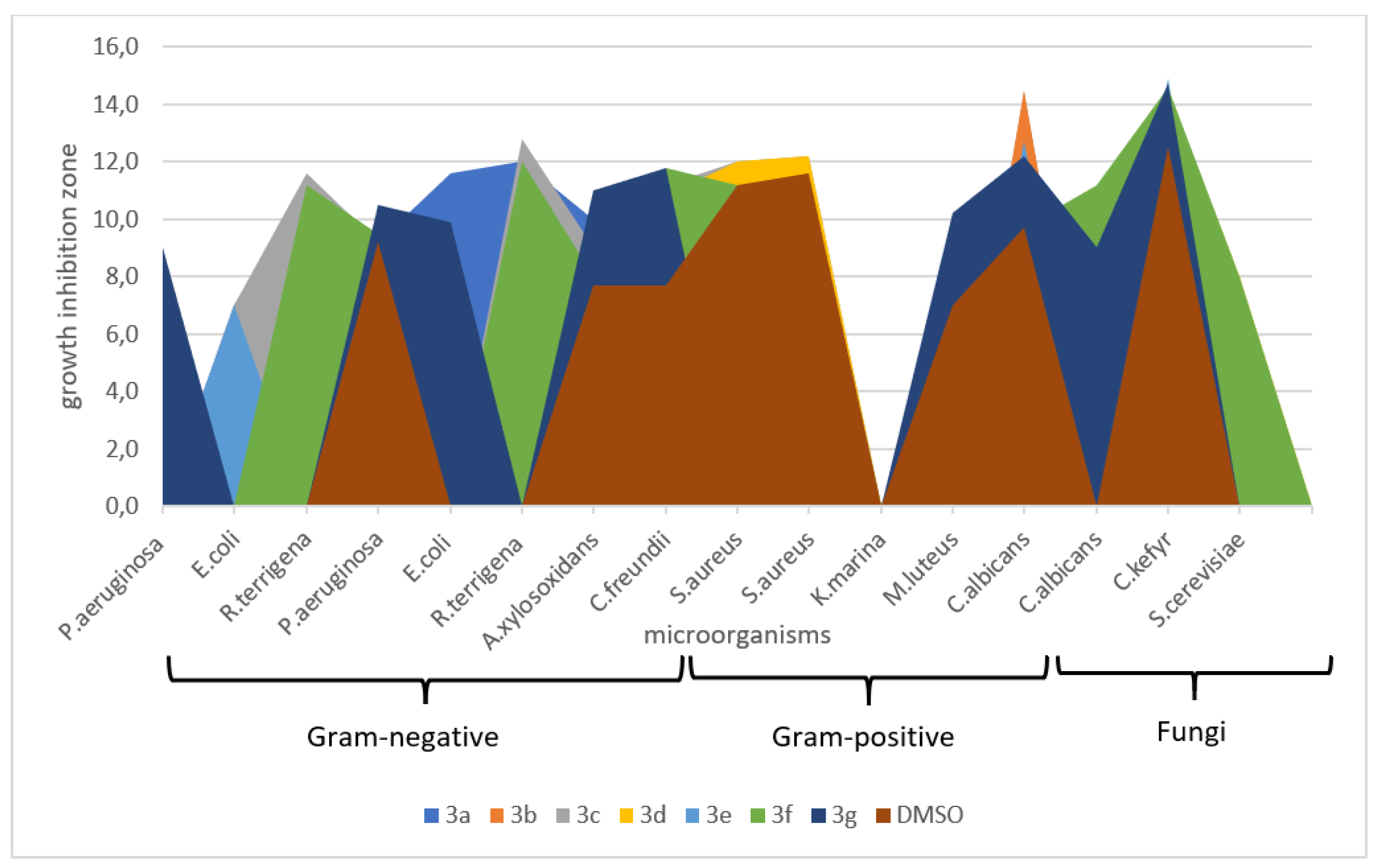
2.3. Antimicrobial Activity
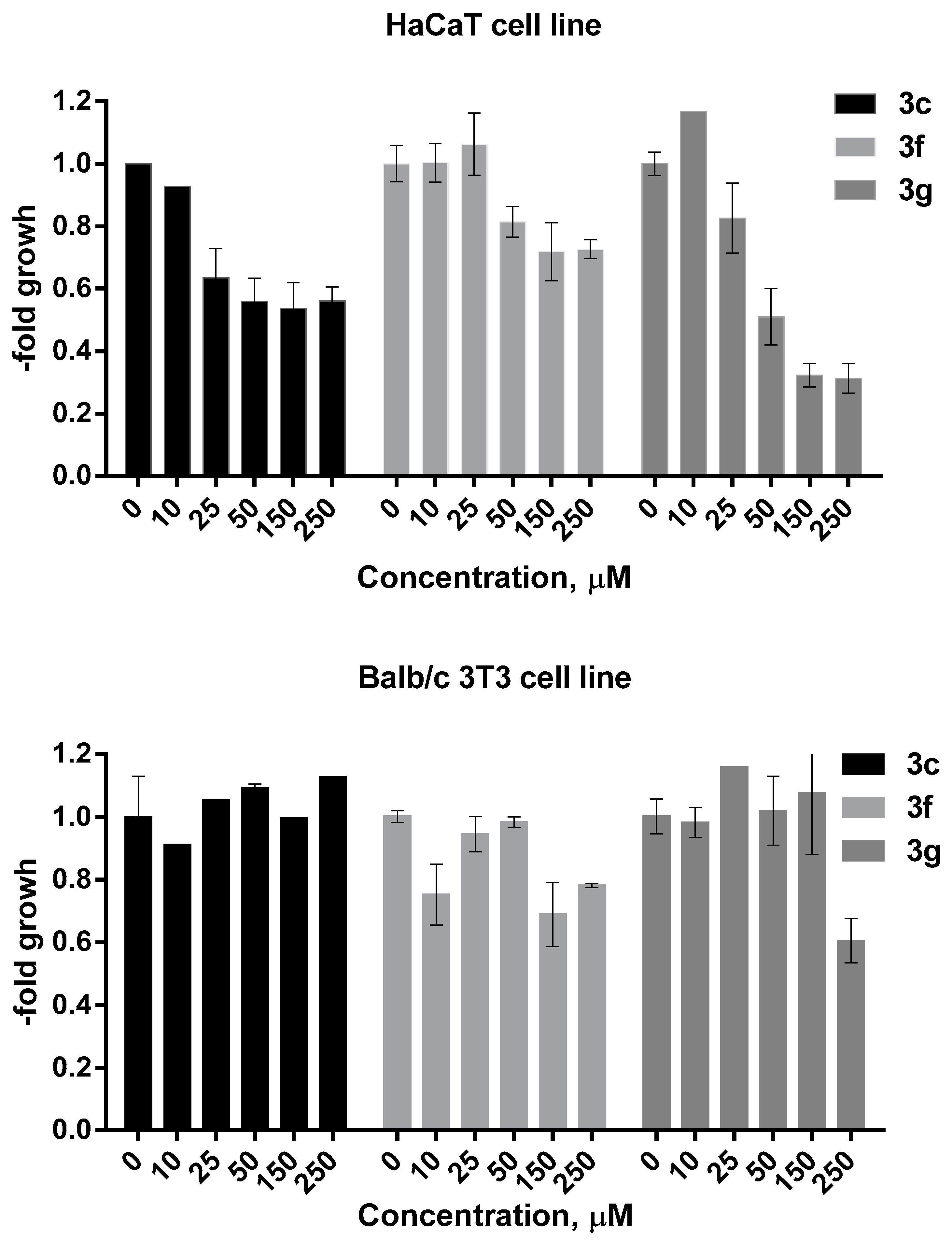
2.4. MTT Assay
2.5. Molecular Docking
3. Results and Discussion
3.1. Synthesis
3.2. Biological Activity
3.3. Molecular and Pharmacokinetic Properties
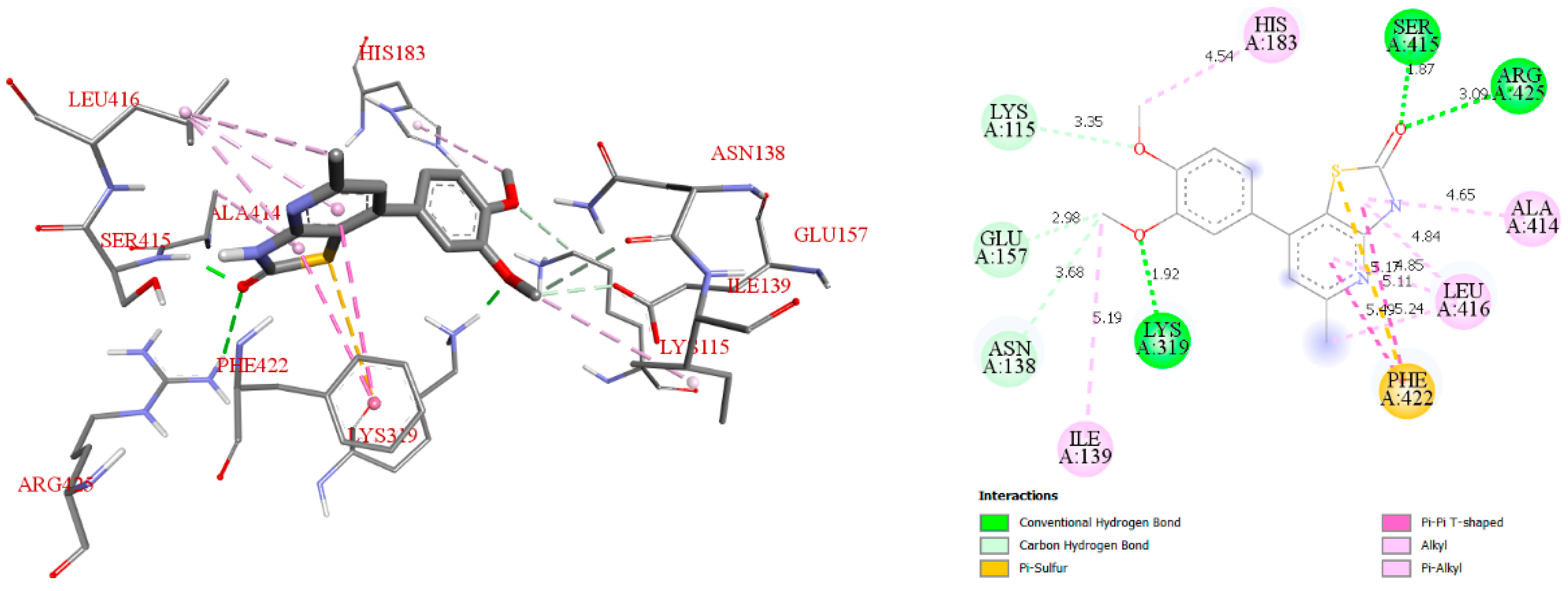
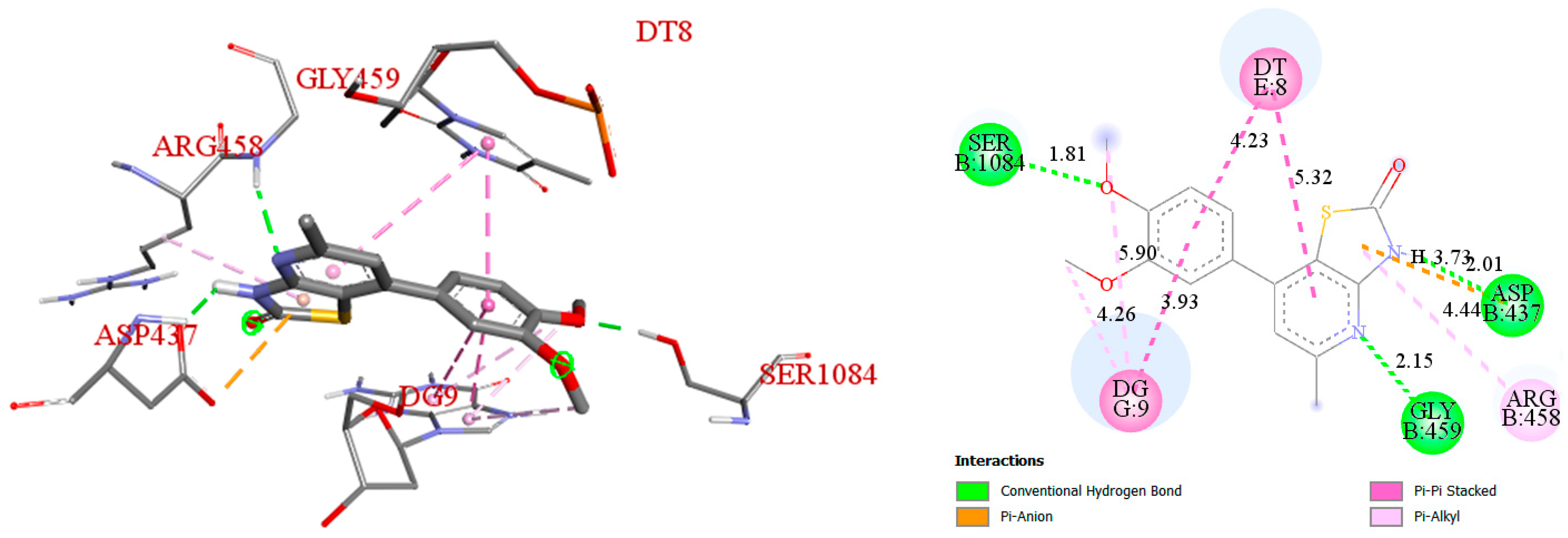
3.4. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lukevits, E. Pyridine derivatives in the drug arsenal (150 years of pyridine chemistry). Chem. Heterocycl. Compd. 1995, 31, 639–650. [Google Scholar] [CrossRef]
- Kishbaugh, T.L.S. Pyridines and Imidazopyridines with medicinal significance. Curr. Top. Med. Chem. 2016, 16, 3274–3302. [Google Scholar] [CrossRef]
- Damani, L.A.; Crooks, P.A.; Shaker, M.S.; Caldwell, J.; D’souza, J.; Smith, R.L. Species differences in the metabolic C-and N-oxidation, and N-methylation of [14C] pyridine in vivo. Xenobiotica 1982, 12, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Carlson, G.P. Comparison of the effects of pyridine and its metabolites on rat liver and kidney. Toxicol. Lett. 1996, 85, 173–178. [Google Scholar] [CrossRef]
- Cronin, M.T.D.; Dearden, J.C.; Duffy, J.C.; Edwards, R.; Manga, N.; Worth, A.P.; Worgan, A.D.P. The importance of hydrophobicity and electrophilicity descriptors in mechanistically-based QSARs for toxicological endpoints. SAR QSAR Environ. Res. 2002, 13, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.S.; Ali, M.A.M. Novel pyrazolo[3,4-b]pyridine derivatives: Synthesis, characterization, antimicrobial and antiproliferative profile. Biol. Pharm. Bull. 2016, 39, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Al-Tel, T.H.; Al-Qawasmeh, R.A.; Zaarour, R. Design, synthesis and in vitro antimicrobial evaluation of novel Imidazo[1,2-a]pyridine and imidazo[2,1-b][1,3]benzothiazole motifs. Eur. J. Med. Chem. 2011, 46, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Arabshahi, H.J.; Van Rensburg, M.; Pilkington, L.I.; Jeon, C.Y.; Song, M.; Gridel, L.M.; Leung, E.; Barker, D.; Vuica-Ross, M.; Volcho, K.P.; et al. A synthesis, in silico, in vitro and in vivo study of thieno[2,3-b]pyridine anticancer analogues. MedChemComm 2015, 6, 1987–1997. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Naggar, A.M.; El-Hashash, M.A. Design, synthesis, molecular modeling and biological evaluation of novel 1H-pyrazolo[3,4-b]pyridine derivatives as potential anticancer agents. Bioorg. Chem. 2016, 67, 43–56. [Google Scholar] [CrossRef]
- Dias, L.R.; Santos, M.B.; de Albuquerque, S.; Castro, H.C.; de Souza, A.M.; Freitas, A.C.; DiVaio, M.A.; Cabral, L.M.; Rodrigues, C.R. Synthesis, in vitro evaluation, and SAR studies of a potential antichagasic 1H-pyrazolo[3,4-b]pyridine series. Bioorg. Med. Chem. 2007, 15, 211–219. [Google Scholar] [CrossRef]
- Shi, F.; Li, C.; Xia, M.; Miao, K.; Zhao, Y.; Tu, S.; Zheng, W.; Zhang, G.; Ma, N. Green chemoselective synthesis of thiazolo[3,2-a]pyridine derivatives and evaluation of their antioxidant and cytotoxic activities. Bioorg. Med. Chem. Lett. 2009, 19, 5565–5568. [Google Scholar] [CrossRef] [PubMed]
- Al-Omar, M.A.; Youssef, K.M.; El-Sherbeny, M.A.; Awadalla, S.A.A.; El-Subbagh, H.I. Synthesis and in vitro antioxidant activity of some new fused pyridine analogs. Arch. Pharm. 2005, 338, 175–180. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Azouz, A.A. Novel pyrimidine-pyridine hybrids: Synthesis, cyclooxygenase inhibition, anti-inflammatory activity and ulcerogenic liability. Bioorg. Chem. 2018, 77, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.T.; Huang, W.H.; Huang, C.K.; Liu, F.C.; Huang, R.Y.; Wu, C.C.; Lee, A.R. Synthesis and anti-inflammatory activities of 4H-chromene and chromeno[2,3-b]pyridine derivatives. Res. Chem. Intermed. 2016, 42, 1195–1215. [Google Scholar] [CrossRef]
- Witherington, J.; Bordas, V.; Garland, S.L.; Hickey, D.M.; Ife, R.J.; Liddle, J.; Saunders, M.; Smith, D.G.; Ward, R.W. 5-Aryl-pyrazolo[3,4-b]pyridines: Potent inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2003, 13, 1577–1580. [Google Scholar] [CrossRef]
- Chioua, M.; Samadi, A.; Soriano, E.; Lozach, O.; Meijer, L.; Marco-Contelles, J. Synthesis and biological evaluation of 3,6-diamino-1H-pyrazolo[3,4-b]pyridine derivatives as protein kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4566–4569. [Google Scholar] [CrossRef] [PubMed]
- Othman, I.M.; Gad-Elkareem, M.A.; Snoussi, M.; Aouadi, K.; Kadri, A. Novel fused pyridine derivatives containing pyrimidine moiety as prospective tyrosyl-tRNA synthetase inhibitors: Design, synthesis, pharmacokinetics and molecular docking studies. J. Mol. Struct. 2020, 1219, 128651. [Google Scholar] [CrossRef]
- Sun, Q.Z.; Lin, G.F.; Li, L.L.; Jin, X.T.; Huang, L.Y.; Zhang, G.; Yang, W.; Chen, K.; Xiang, R.; Chen, C.; et al. Discovery of potent and selective inhibitors of cdc2-like kinase 1 (clk1) as a new class of autophagy inducers. J. Med. Chem. 2017, 60, 6337–6352. [Google Scholar] [CrossRef]
- Fujita, M.; Seki, T.; Ikeda, N. Synthesis and bioactivities of novel bicyclic thiophenes and 4,5,6,7-tetrahydrothieno[2,3-c]pyridines as inhibitors of tumor necrosis factor-α (TNF-α) production. Bioorg. Med. Chem. Lett. 2002, 12, 1897–1900. [Google Scholar] [CrossRef]
- Das, D.; Sikdar, P.; Bairagi, M. Recent developments of 2-aminothiazoles in medicinal chemistry. Eur. J. Med. Chem. 2016, 109, 89–98. [Google Scholar] [CrossRef]
- Bondock, S.; Albormani, O.; Fouda, A.M.; Abu Safieh, K.A. Progress in the chemistry of 5-acetylthiazoles. Synth. Commun. 2016, 46, 1081–1117. [Google Scholar] [CrossRef]
- Chebanov, V.A.; Saraev, V.E.; Desenko, S.M.; Chernenko, V.N.; Knyazeva, I.V.; Groth, U.; Glasnov, T.N.; Kappe, C.O. Tuning of chemo-and regioselectivities in multicomponent condensations of 5-aminopyrazoles, dimedone, and aldehydes. J. Org. Chem. 2008, 73, 5110–5118. [Google Scholar] [CrossRef] [PubMed]
- Chebanov, V.A.; Sakhno, Y.I.; Desenko, S.M.; Chernenko, V.N.; Musatov, V.I.; Shishkina, S.V.; Shishkin, O.V.; Kappe, C.O. Cyclocondensation reactions of 5-aminopyrazoles, pyruvic acids and aldehydes. Multicomponent approaches to pyrazolopyridines and related products. Tetrahedron 2007, 63, 1229–1242. [Google Scholar] [CrossRef]
- Chebanov, V.A.; Desenko, S.M. Dihydroazines based on α,β-unsaturated ketones reactions. Curr. Org. Chem. 2006, 10, 297. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zimenkovsky, B.; Radko, L.; Stypula-Trebas, S.; Roman, O.; Gzella, A.K.; Lesyk, R. Synthesis and cytotoxicity of new thiazolo[4,5-b]pyridine-2(3H)-one derivatives based on α,β-unsaturated ketones and α-ketoacids. Chem. Pap. 2018, 72, 669–681. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zimenkovsky, B.; Gzella, A.K.; Lesyk, R. Arylidene pyruvic acids motif in the synthesis of new 2H,5H-chromeno [4′,3′:4,5]thiopyrano[2,3-d]thiazoles via tandem hetero-Diels–Alder-hemiacetal reaction. Synth. Commun. 2015, 45, 2266–2270. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zimenkovsky, B.; Nektegayev, I.; Lesyk, R. Arylidene pyruvic acids motif in the synthesis of new thiopyrano[2,3-d] thiazoles as potential biologically active compounds. Heterocycl. Commun. 2015, 21, 55–59. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zasidko, V.; Atamanyuk, D.; Kaminskyy, D.; Derkach, H.; Karpenko, O.; Ogurtsov, V.; Kutsyk, R.; Lesyk, R. Synthesis, antioxidant and antimicrobial activities of novel thiopyrano[2,3-d]thiazoles based on aroylacrylic acids. Mol. Divers. 2017, 21, 427–436. [Google Scholar] [CrossRef]
- Khodarahmi, G.; Asadi, P.; Farrokhpour, H.; Hassanzadeh, F.; Dinari, M. Design of novel potential aromatase inhibitors via hybrid pharmacophore approach: Docking improvement using the QM/MM method. RSC Adv. 2015, 5, 58055–58064. [Google Scholar] [CrossRef]
- Chaban, T.I.; Ogurtsov, V.V.; Chaban, I.G.; Klenina, O.V.; Komarytsia, J.D. Synthesis and antioxidant activity evaluation of novel 5,7-dimethyl-3H-thiazolo[4,5-b]pyridines. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1611–1620. [Google Scholar] [CrossRef]
- Chaban, T.I.; Ogurtsov, V.V.; Matiychuk, V.S.; Chaban, I.G.; Demchuk, I.L.; Nektegayev, I.A. Synthesis, anti-inflammatory and antioxidant activities of novel 3H-thiazolo[4,5-b]pyridines. Acta Chim. Slov. 2019, 66, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gaby, M.S.A.; Al-Sehemi, A.G.; Mohamed, Y.A.; Ammar, Y.A. Recent trends in chemistry of thiazolopyridines. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 631–674. [Google Scholar] [CrossRef]
- Hegde, S.G.; Mahoney, M.D. Synthesis and herbicidal activity of 5-(haloalkyl)-substituted thiazolo[4,5-b]pyridine-3(2H)-acetic acid derivatives. J. Agric. Food Chem. 1993, 41, 2131–2134. [Google Scholar] [CrossRef]
- Lozynskyi, A.; Zimenkovsky, B.; Ivasechko, I.; Senkiv, J.; Gzella, A.; Karpenko, O.; Stoika, R.; Lesyk, R. Synthesis and cytotoxicity of new 2-oxo-7-phenyl-2,3-dihydrothiazolo[4,5-b]pyridine-5-carboxylic acid amides. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 1149–1157. [Google Scholar] [CrossRef]
- Lozynskyi, A.V.; Derkach, H.O.; Zasidko, V.V.; Konechnyi, Y.T.; Finiuk, N.S.; Len, Y.T.; Kutsyk, R.V.; Regeda, M.S.; Lesyk, R.B. Antimicrobial and cytotoxic activities of thiazolo[4,5-b]pyridine derivatives. Biopolym. Cell 2021, 37, 153. [Google Scholar] [CrossRef]
- Komaritsa, I.D. Studies on azolidones and their derivatives. Chem. Heterocycl. Compd. 1968, 4, 324–325. [Google Scholar] [CrossRef]
- Skrobiszewski, A.; Ogórek, R.; Pląskowska, E.; Gładkowski, W. Pleurotus ostreatus as a source of enoate reductase. Biocatal. Agric. Biotechnol. 2013, 2, 26–31. [Google Scholar] [CrossRef]
- EUCAST. Disk Diffusion—Manual v 8.0 (1 January 2020). Available online: http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/2020_manuals/Manual_v_8.0_EUCAST_Disk_Test_2020.pdf (accessed on 20 July 2021).
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Tomašić, T.; Zidar, N.; Rupnik, V.; Kovač, A.; Blanot, D.; Gobec, S.; Kikelj, D.; Mašič, L.P. Synthesis and biological evaluation of new glutamic acid-based inhibitors of MurD ligase. Bioorg. Med. Chem. Lett. 2009, 19, 153–157. [Google Scholar] [CrossRef]
- Tomasic, T.; Katsamakas, S.; Hodnik, Z.; Ilaš, J.; Brvar, M.; Solmajer, T.; Montalvão, S.; Tammela, P.; Banjanac, M.; Ergović, G.; et al. Discovery of 4, 5, 6, 7-tetrahydrobenzo[1,2-d]thiazoles as novel DNA gyrase inhibitors targeting the ATP-binding site. J. Med. Chem. 2015, 58, 5501–5521. [Google Scholar] [CrossRef]
- Zidar, N.; Tomašić, T.; Šink, R.; Kovač, A.; Patin, D.; Blanot, D.; Contreras-Martel, C.; Dessen, A.; Premru, M.M.; Zega, A.; et al. New 5-benzylidenethiazolidin-4-one inhibitors of bacterial MurD ligase: Design, synthesis, crystal structures, and biological evaluation. Eur. J. Med. Chem. 2011, 46, 5512–5523. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Aiken, A.M.; Allegranzi, B.; Scott, J.A.; Mehtar, S.; Pittet, D.; Grundmann, H. Antibiotic resistance needs global solutions. Lancet Infect. Dis. 2014, 14, 550–551. [Google Scholar] [CrossRef]
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 27 March 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Species | Species of Bacteria and Fungi | Zone of Growth Inhibition (mm ± SE) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 3d | 3e | 3f | 3g | DMSO | Vancomycin | Ciprofloxacin | Clotrimazole | ||||
| 1 | Gram-negative bacteria | Reference strains | Pseudomonas aeruginosa (ATCC 27853 (F-51)) | 00 | 00 | 00 | 00 | 00 | 7.0 ± 0.3 ** | 9.0 ± 0.4 ** | 00 | - | 35.0 ± 0.3 | - |
| 2 | Escherichia coli (ATCC 25922) | 7.0 ± 0.25 ** | 00 | 7.0 ± 0.3 ** | 00 | 7.0 ± 0.2 ** | 00 | 00 | 00 | - | 42.0 ± 0.5 | - | ||
| 3 | Raoultella terrigena (ATCC 33257) | 11.0 ± 0.4 ** | 00 | 11.6 ± 0.3 ** | 00 | 00 | 11.2 ± 0.4 ** | 00 | 00 | - | 30.0 ± 0.5 | - | ||
| 4 | Clinical strains | Pseudomonas aeruginosa N 7 | 9.2 ± 0.2 | 9.2 ± 0.2 | 9.2 ± 0.3 ** | 9.2 ± 0.2 | 9.2 ± 0.2 | 9.5 ± 0.3 | 10.5 ± 0.4 * | 9.2 ± 0.2 | - | 20.0 ± 0.2 | - | |
| 5 | Escherichia coli N 37 | 11.6 ± 0.25 ** | 00 | 9.2 ± 0.2 ** | 00 | 00 | 00 | 9.9 ± 0.2 ** | 00 | - | 14.0 ± 0.4 | - | ||
| 6 | Raoultella terrigena N1 | 12.0 ± 0.5 ** | 00 | 00 | 00 | 00 | 12.0 ± 0.3** | 00 | 00 | - | 12.0 ± 0.3 | - | ||
| 7 | Achromobacter xylosoxidans N 147 | 10.0 ± 0.4 ** | 8.0 ± 0.3 | 12.8 ± 0.3 ** | 10.0 ± 0.4 ** | 8.0 ± 0.3 | 8.0 ± 0.3 | 11.0 ± 0.2 ** | 8.0 ± 0.2 | - | 15.0 ± 0.5 | - | ||
| 8 | Citrobacter freundii N 2 | 7.7 ± 0.2 | 7.7 ± 0.2 | 9.0 ± 0.5 * | 10.8 ± 0.4 ** | 7.7 ± 0.2 | 11.8 ± 0.4** | 11.8 ± 0.5 ** | 7.7 ± 0.2 | - | 16.0 ± 0.3 | - | ||
| 9 | Gram-positive bacteria | Reference strain | Staphylococcus aureus (ATCC 25923 (F-49)) | 00 | 00 | 11.2 ± 0.5 | 12.0 ± 0.3 | 00 | 11.2 ± 0.3 | 00 | 11.2 ± 0.4 | 32 ± 0.5 | 35.0 ± 0.5 | - |
| 10 | Clinical strains | Staphylococcus aureus N 23 | 00 | 00 | 12.0 ± 0.3 | 12.2 ± 0.4 | 00 | 11.4 ± 0.3 | 00 | 11.6 ± 0.2 | 11.4 ± 0.3 | 9.0 ± 0.2 | - | |
| 11 | Kocuria marina N 133 | 00 | 00 | 12.2 ± 0.4 ** | 00 | 00 | 00 | 00 | 00 | 25.1 ± 0.4 | 8.0 ± 0.2 | - | ||
| 12 | Micrococcus luteus N 136 | 00 | 00 | 00 | 00 | 00 | 00 | 10.2 ± 0.4 ** | 7.0 ± 0.3 | 12.0 ± 0.2 | 10.0 ± 0.2 | - | ||
| 13 | Fungi | Reference strain | Candida. albicans (ATCC 885-653) | 14.4 ± 0.4 ** | 14.5 ± 0.5 ** | 00 | 9.6 ± 0.3 | 12.7 ± 0.4 ** | 9.7 ± 0.3 | 12.2 ± 0.2 | 9.7 ± 0.2 ** | - | - | 18.0 ± 0.5 |
| 14 | Clinical strains | Candida albicans N 60 | 00 | 00 | 9.7 ± 0.3 | 9.2 ± 0.4 ** | 00 | 11.2 ± 0.5 ** | 9.0 ± 0.3 ** | 00 | - | - | 11.0 ± 0.3 | |
| 15 | Candida kefyr N 68 | 13.8 ± 0.1 ** | 12.3 ± 0.4 | 9.8 ± 0.4 ** | 14.5 ± 0.4 ** | 14.9 ± 0.3 ** | 14.6 ± 0.2 ** | 14.7 ± 0.2 ** | 12.5 ± 0.2 | - | - | 6.0 ± 0.2 | ||
| 16 | Saccharomyces cerevisiae N 62 | 00 | 00 | 12.5 ± 0.4 ** | 8,0 ± 0.2 ** | 00 | 8,0 ± 0.2 ** | 00 | 00 | - | - | 8.0 ± 0.2 | ||
| μM | |||||||
|---|---|---|---|---|---|---|---|
| 3a | 3c | 3f | 3g | Vancomycin | Ciprofloxacin | Clotrimazole | |
| Pseudomonas aeruginosa N 7 | - | - | 1.56 | 0.21 | - | 1.81 | - |
| Escherichia coli N 37 | 1.84 | 0.96 | - | 0.21 | - | 1.51 | - |
| Raoultella terrigena N1 | 1.84 | 0.96 | 0.78 | - | - | 2.11 | - |
| Citrobacter freundii N 2 | - | 0.96 | 1.56 | 1.66 | - | 2.26 | - |
| Staphylococcus aureus N 23 | - | 0.96 | - | - | 4.16 | 3.02 | - |
| Micrococcus luteus N 136 | - | - | - | 1.66 | 2.77 | 4.53 | - |
| Candida. albicans (ATCC 885-653) | 1.84 | 0.96 | 1.56 | 0.83 | - | - | 1.16 |
| Candida albicans N 60 | 1.84 | 1.92 | 1.56 | 1.66 | - | - | 2.9 |
| 3c | 3f | 3g | |
|---|---|---|---|
| Physicochemical properties | |||
| Molecular weight | 260.29 | 321.19 | 302.35 |
| Num. heavy atoms | 18 | 18 | 21 |
| Num. arom. heavy atoms | 15 | 15 | 15 |
| Num. rotatable bonds | 1 | 1 | 3 |
| Num. H-bond acceptors | 3 | 2 | 4 |
| Num. H-bond donors | 1 | 1 | 1 |
| Molar Refractivity | 70.60 | 78.34 | 83.63 |
| TPSA Ų | 73.99 | 73.99 | 92.45 |
| Consensus log Po/w | 3.35 | 3.67 | 2.99 |
| Lipinski’Rule | Yes | Yes | Yes |
| Pharmacokinetics | |||
| GI absorption | High | High | High |
| BBB permeant | Yes | Yes | No |
| p-gp substrate | No | No | No |
| Log Kp (SP) (cm/s) (skin permeation) | −5.71 | −5.66 | −6.08 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 |
| MurD (2Y67) | DNA Gyrase (2XCT) | |||
|---|---|---|---|---|
| COMPOUND | Estimated Free Energy of Binding (kcal/mol) | Estimated Inhibitory Constant, Ki (μM) | Estimated Free Energy of Binding (kcal/mol) | Estimated Inhibitory Constant, Ki |
| 3g | −6.31 | 23.63 uM | −8.28 | 858.77 nM |
| Ciprofloxacin | - | - | −8.35 | 756.11 nM |
| N21 | −6.08 | 34.80 uM | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozynskyi, A.; Konechnyi, Y.; Senkiv, J.; Yushyn, I.; Khyluk, D.; Karpenko, O.; Shepeta, Y.; Lesyk, R. Synthesis and Biological Activity Evaluation of Novel 5-Methyl-7-phenyl-3H-thiazolo[4,5-b]pyridin-2-ones. Sci. Pharm. 2021, 89, 52. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm89040052
Lozynskyi A, Konechnyi Y, Senkiv J, Yushyn I, Khyluk D, Karpenko O, Shepeta Y, Lesyk R. Synthesis and Biological Activity Evaluation of Novel 5-Methyl-7-phenyl-3H-thiazolo[4,5-b]pyridin-2-ones. Scientia Pharmaceutica. 2021; 89(4):52. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm89040052
Chicago/Turabian StyleLozynskyi, Andrii, Yulian Konechnyi, Julia Senkiv, Ihor Yushyn, Dmytro Khyluk, Olexandr Karpenko, Yulia Shepeta, and Roman Lesyk. 2021. "Synthesis and Biological Activity Evaluation of Novel 5-Methyl-7-phenyl-3H-thiazolo[4,5-b]pyridin-2-ones" Scientia Pharmaceutica 89, no. 4: 52. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm89040052