Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine
by
 , and
, and
Haibo Mei
1 ,
,
Zizhen Yin
1,
Toshio Miwa
2,
Hiroki Moriwaki
2,*,
Hidenori Abe
2,
Jianlin Han
1,* and
Vadim A. Soloshonok
3,4,* 1
Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China
2
Hamari Chemicals Ltd., 1-4-29 Kunijima, Higashi-Yodogawa-ku, Osaka 533-0024, Japan
3
Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, 20018 San Sebastián, Spain
4
IKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, Plaza Bizkaia, 48013 Bilbao, Spain
*
Authors to whom correspondence should be addressed.
Symmetry 2019, 11(4), 578; https://0-doi-org.brum.beds.ac.uk/10.3390/sym11040578
Submission received: 8 April 2019
/
Revised: 16 April 2019
/
Accepted: 18 April 2019
/
Published: 21 April 2019
(This article belongs to the Special Issue Amino Acid Chirality)
Abstract
:In this work we report a convenient asymmetric synthesis of Fmoc-(S)-6,6,6-trifluoro-norleucine via alkylation reaction of chiral glycine equivalent. The target amino acid of 99% enantiomeric purity was prepared with 82.4% total yield (three steps).
1. Introduction
Medicinal applications of new organic compounds have always been a major driving force behind the development of organic methodology. In this regard, one can notice two general trends in the design of modern pharmaceutical drugs: the introduction of fluorine-containing substituents and tailor-made amino acids (AAs) [1,2,3,4,5,6,7,8,9,10]. While the strategic fluorination usually leads to improved pharmacokinetics and greater oxidative metabolic stability [9,11,12], the presence of tailor-made amino AAs residues allows for more precise mimicking of the natural peptide–receptor interactions [1,2,3,4,5,6,7,8]. Subsequently, fluorine-containing α- [13,14,15,16,17,18,19,20,21] and β-AAs [22,23,24], featuring both structural traits, are currently an increasingly important class of compounds used in bio-medicinal studies and drug design [25,26,27,28]. For example, (S)-2-amino-6,6,6-trifluorohexanoic acid and 6,6,6-trifluoro-norleucine 1 (Scheme 1) and its derivatives were shown to possess interesting biological properties, such as antitumor [29], antimicrobial [30], and enzyme inhibitory activity [31,32]. However, the major interest in fluorinated AA 1 is related to its various applications in the therapeutic peptide engineering [33,34,35,36] and protein structural studies [37,38,39]. Over the last decade, various synthetic approaches for preparation of tailor-made fluorinated norleucine 1 have received due attention. One group of the methods is based on elaboration of functional groups in the already prearranged AA skeleton 2, such as additions of CF3 radical to the terminal C=C bond [40,41] or biomimetic transamination [42,43]. However, a more general approach for synthesis of AA 1 includes alkyl halide alkylation of properly protected glycine derivatives 3 [44,45,46,47,48]. These reactions can be conducted under homogeneous [45,46,47,48], as well as PTC conditions [44].
In this paper we describe a convenient asymmetric synthesis of Fmoc derivative 6,6,6-trifluoro-norleucine 1 via CF3(CH2)3I alkylation of the recently rationally designed chiral equivalent of nucleophilic glycine 4. The method is operationally convenient, robust, scalable, and can be recommended for practical preparation of enantiomerically pure (~99% ee) derivative of this important tailor-made AA.
2. Materials and Methods
General Methods. All solvents and reagents were used as purchased without further purification. All reactions were conducted by magnetically stirring and detected by routine chromatography on TLC plates. Flash chromatography was carried out using the corresponding solvents on silica gel (0.064–0.210 mm). The reported yields are for isolated and chemically pure compounds. HPLC experiments were performed on a standard equipment using the InertsilTM ODS-3 column (3 μm, 150 × 4.6 mm) ran at 1.0 mL/min, 30 °C; monitoring was set at 254 nm with a gradient of 10 mM aqueous HCOOH/NH3 containing 0.1% HCOOH (eluent A) and MeCN (eluent B) from A: B = 95:5 to 20:80 and 20:80. 1H-, 19F-, and 13C-NMR data were recorded on Bruker AVANCE III-400 instrument. Chemical shifts are presented in ppm (d), referenced to SiMe4 (TMS). Optical rotations data were conducted on a DIP-370 instrument. Melting points were taken as usual.
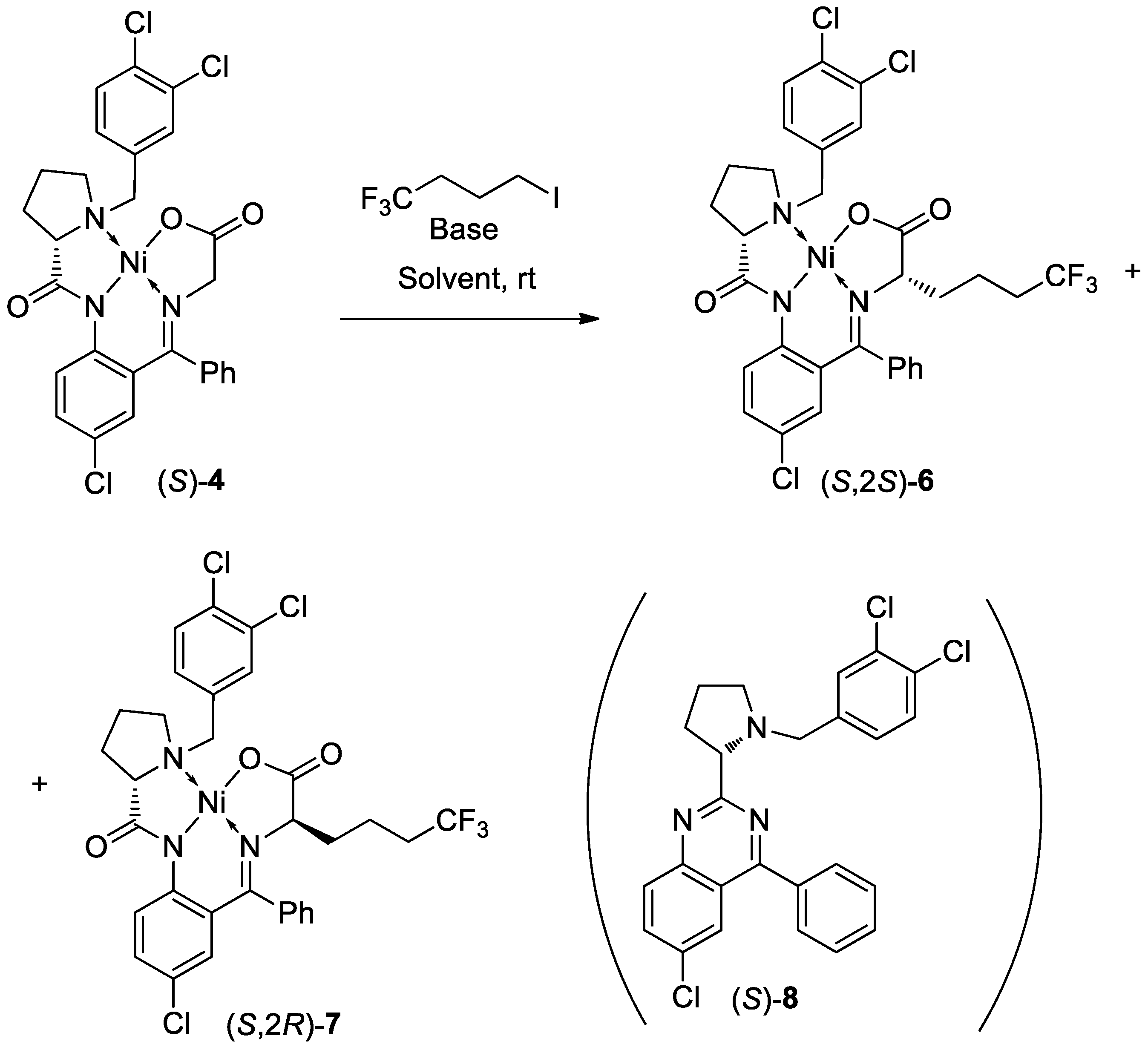
Alkylation Reaction of Glycine Complex (S)-4 with CF3(CH2)3I. The Ni–glycine complex (S)-4 (20.0 g, 33.2 mmol, 1.0 equiv.) and 1,1,1-trifluoro-4-iodobutane (7.90 g, 33.2 mmol, 1.0 equiv.) were stirred in deoxygenated N,N-dimethyl-formamide (DMF) (140 mL, 7 v/w) at room temperature under argon. Then, 10% NaOMe methanol solution (1.0 equiv.) was added into the above mixture. The resulting solution was stirred at room temperature for 2 h, and then was poured into water (46 mL) at same temperature to give the precipitate. After 0.5 h, the mixture was added water (24 mL), and was stirred for 15 h. After that, the precipitate was filtered, washed with DMF-H2O (36 mL, 2:1 v/v), washed with water (40 mL) and dried in vacuo at 60 °C for 7 h to afford the crude Ni complex (20.8 g, 87.9%, a red solid) as a mixture of (S,2S)-6 and (S,2R)-7, the diastereomeric ratio was determined to be (98.7 %de) by HPLC analysis, in which the major (S,2S)-6 was eluted at a retention time (tR) of 20.3 min and the minor (S,2R)-7 at 21.5 min under the conditions described in the general methods. The mixture of 6 and 7 was purified by column to give the diastereomerically pure major product 6 in 87.5% yield (see Supplementary Materials).
(S,2S)-6 (major isomer): M.p. 229–231 °C. [α]25D = +2616 (c = 0.2, CH3OH). 1H NMR (400 MHz, CDCl3): δ = 8.89 (d, J = 1.6 Hz, 1H), 8.09 (d, J = 9.2 Hz, 1H), 7.77 (dd, J = 1.7, 8.1 Hz, 1H), 7.49–7.58 (m, 3H), 7.37 (d, J = 8.1 Hz, 1H), 7.29–7.30 (m, 1H), 7.11 (dd, J = 2.4, 9.2 Hz, 1H), 6.88 (d, J = 7.5 Hz, 1H), 6.59 (d, J = 2.4 Hz, 1H), 4.34 (d, J = 12.6 Hz, 1H), 3.87 (dd, J = 8.0, 3.4 Hz, 1H), 3.51–3.57 (m, 2H), 3.35–3.39 (m, 1H), 3.21 (d, J = 12.6 Hz, 1H), 2.59–2.71 (m, 2H), 2.35–2.37 (m, 1H), 2.24–2.25 (m, 1H), 1.84–2.08 (m, 3H), 1.82–1.84 (m, 2H), 1.60–1.66 (m, 1H). 13C NMR (100 MHz, CDCl3): δ = 18.0, 23.5, 30.8 32.6 (J = 29.0 Hz, q), 34.1, 58.3, 63.0, 69.7, 71.3, 124.1, 125.3 (J = 276.6 Hz, q), 125.7, 127.0, 127.1, 127.2, 129.3, 129.4, 129.8, 130.3, 132.1, 132.4, 132.7, 133.3, 133.5, 133.6, 134.8, 140.5, 170.4, 178.3, 179.9. 19F NMR (376 MHz, CDCl3): δ = −66.8 (CF3). IR (KBr): ν = 2977, 1674, 1650, 1535, 1463, 1398, 1251, 1188, 1077, 826 cm−1. MS (ESI): m/z = 710.1 [M + H]+.
(S,2R)-7 (minor isomer): M.p. 218–220 °C. [α]25D= −1998 (c = 0.2, CH3OH). 1H NMR (400 MHz, CDCl3): δ = 8.50 (d, J = 9.2 Hz, 1H), 8.37 (d, J = 2.0 Hz, 1H), 7.74 (dd, J = 2.0, 8.0 Hz, 1H), 7.03–7.23 (m, 4H), 7.20–7.23 (m, 2H), 7.00–7.03 (m, 1H), 6.72 (d, J = 2.4, 1H), 4.24–4.30 (m, 2H), 3.71 (dd, J = 10.0, 3.2 Hz, 1H), 3.55 (dd, J = 8.8, 4.4 Hz, 1H), 3.35–3.38 (m, 1H), 2.62–2.67 (m, 2H), 2.25–2.30 (m, 2H), 1.80–2.00 (m, 2H), 1.64–1.79 (m, 3H), 1.25–1.35 (m, 2H). 13C NMR (100 MHz, CDCl3): δ = 18.3, 23.1, 30.3 32.2 (J = 29.0 Hz, q), 35.1, 59.5, 60.5, 69.2, 69.7, 125.0, 125.4 (J = 276.6 Hz, q), 125.7, 126.6, 126.7, 127.6, 129.1, 129.5, 130.2, 130.5, 132.3, 132.5, 133.2, 133.3, 133.4, 133.8, 133.9, 141.3, 170.9, 178.9, 181.5. 19F NMR (376 MHz, CDCl3): δ = −66.6 (CF3). IR (KBr): ν = 2945, 1676, 1644, 1584, 1464, 1395, 1247, 1135, 1029, 823 cm−1.
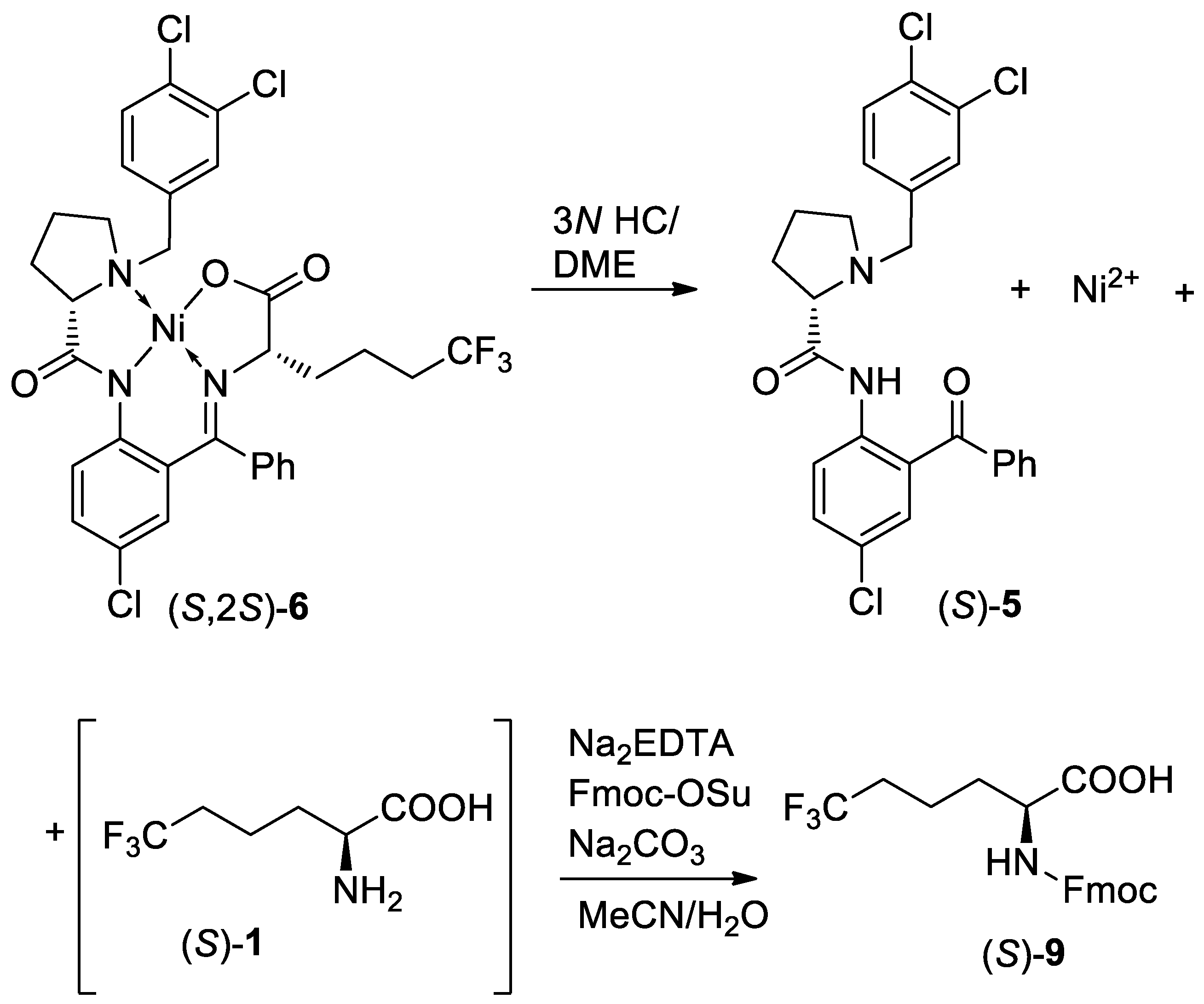
Preparation of Fmoc-(S)-2-amino-6,6,6-trifluorohexanoic acid (S)-9. To a solution of Ni complex (S,2S)-6 (20.0 g, 28.1 mmol, 1.0 equiv.) in dimethoxyethane (DME) (100 mL, 5 v/w) was added HCl (3 N, 46.8 mL, 5.0 equiv.), and the resulting mixture was heated at 50–60 °C for 2 h. Then, the reaction mixture was cooled to room temperature, and the the reaction mixture was evaporated to remove DME. Water (400 mL) was added, and white precipitate (HCl salt) appeared. The precipitate was filtered, washed with water (20 mL × 2). The filtrate was total 80 mL.
To the above (S)-6,6,6-trifluoro-norleucine HCl green solution were added ethylenediaminetetraacetic acid disodium salt hydrate (10.5 g, 1.0 equiv) and acetonitrile (60 mL) and the mixture was stirred for 0.5 h at room temperature; 48% NaOH (9.5 g, 4.1 equiv) was added. Then, sodium carbonate (3.87 g, 1.3 equiv) and Fmoc-OSu (9.48 g, 1.0 equiv.) were added to the resulting mixture. The mixture was stirred for 3 h at room temperature, and then was concentrated. To the residue was added ethyl acetate (100 mL) and HCl (6N, 20.0 mL), and the phases were separated. The aqueous layer was extracted with ethyl acetate (40 mL) and the combined organic layer was washed with water (40 mL) and 10 % brine (40 mL). The organic solution was dried with Na2SO4, and then the filtrate was concentrated to dryness and dried in vacuo at 50 °C to afford (S)-9 (11.45 g, a white powder) (see Supplementary Materials).
(S)-9: M.p. 152–154 °C. 1H NMR (400 MHz, CD3OD): δ = 7.77–7.79 (m, 2H), 7.64–7.69 (m, 2H), 7.32–7.40 (m, 2H), 7.28–7.30 (m, 2H), 4.36–4.37 (m, 2H), 4.18–4.24 (m, 2H), 2.18–2.23 (m, 2H), 1.92–1.95 (m, 1H), 1.64–1.77 (m, 2H). 19F NMR (376 MHz, CDCl3): δ = –67.6 (CF3). IR (KBr): ν = 3265, 3067, 2926, 2858, 1654, 1476, 1445, 1049 cm−1. MS (ESI): m/z = 431.1 [M + Na]+.
3. Results and Discussion
In line with our longstanding curiosity in synthesis of several types of tailor-made AAs, in particular trifluoromethyl- and [49,50] phosphorus-containing [51,52], sterically constrained [53,54], and nonlinear optical properties of AA and their derivatives, such as self-disproportionation of enantiomers [55,56,57], we were contributing to the chemistry of Ni(II) complexes of Schiff bases of AA as a general methodology for synthesis of tailor-made AAs [58,59,60]. Over the last several years, we were focusing on the modular design [61,62] of chiral tridentate ligands used for preparation of the corresponding Ni(II) complexes of AA Schiff bases. Among other advances [63,64,65], recently we developed a strategically trichloro-substituted ligand 5 (Scheme 2) [66,67], which showed excellent stereocontrolling properties in the dynamic kinetic resolution of unprotected α- [68,69] and β-AAs [70]. It was shown that the presence of strategically positioned chlorine atoms favorably influence the parallel displaced type of aromatic stacking interactions between the proline N-benzyl and o-amino-benzophenone ring [66]. The quality of these aromatic stacking has important synthetic consequences [67] enhancing the stereochemical preferences at the a-position of the amino acid residue. It should be mentioned that the strategically chlorinated ligand 5 was developed by and commercially available from Hamari Chemicals. However, other methodological avenues of ligand 5 applications for the asymmetric preparation of tailor-made AAs still remain unexplored [71]. Ligands (S)- or (R)-5 are commercially available and can be conveniently prepared [72] starting form (S)- or (R)-proline and transformed to the Ni(II) complexes of glycine Schiff base 4 [71,73]. As presented in Scheme 2, ligand 5 is reacting with glycine and Ni(II) ions in basic methanol solution to afford complex 4.
Alkylation of the glycine moiety in complexes of type 4 with alkyl halide as alkyl precursor can be conducted under homogeneous [74] as well as phase-transfer catalysis (PTC) conditions [75]. The latter are usually preferred, due to the low byproducts formation, but can be realized only for activated alkyl halides. Thus, under standard PTC conditions [75], CF3(CH2)3I was found to be totally inefficient for alkylation of complex (S)-4, resulting in noticeable decomposition of the alkylating reagent. In sharp contrast, under the homogeneous conditions (Scheme 3), by use of DMSO as a solvent and NaOH as a base (Table 1), the expected alkylation products were isolated and fully characterized.
HPLC analysis of the reaction mixture revealed rather low rate of the alkylation and disappointing diastereoselectivity. As shown in Table 1, in entries 1–3, after 4.5 h of the reaction time, over 43% of starting complex 4 was still intact. Near-complete consumption of glycine complex 4 was observed after 24 h (entry 4) along with a large amount of various byproducts. One of the major byproducts was identified as previously described [73] compound 8 resulting from oxidative decomposition of stating glycine complex 4 [76,77].
One of the critical notes made in this series of experiment was the observation that solid NaOH is not the best choice of introducing the base into the reaction mixture. After a series of experiments focused on solvent/base issue, we found that combination of DMF as a reaction solvent and solution of NaOMe in MeOH as a base allows for a dramatically improved outcome. Thus, as presented in Table 2, the alkylation of (S)-4 with CF3(CH2)3I performed in DMF and using NaOMe/MeOH (28% solution) proceeded with high rate providing for virtually complete (>99%) consumption of the starting materials within about 30 min (entry 1). Importantly, the amount of byproducts was also dramatically reduced, albeit the stereochemical outcome was rather marginal (90:10 dr). Interestingly, extension of the reaction time from 0.5 to 2.0 h did not result in any visible changes of the chemical or stereochemical outcome (entry 2).
Additional experiments with combination of DMF/NaOMe indicated that the application of less concentrated solution of NaOMe, has some advantageous effect on the reaction outcome. As shown in Table 2 (entry 3), the use of 10% NaOMe solution in MeOH as a base resulted in almost complete alkylation of glycine complex (S)-4 with CF3(CH2)3I in less than 30 min of the reaction time. Similar to the previous experiments (entries 1 and 2) the alkylation proceeded rather cleanly, but most notably with rather improved diastereoselectivity (97:3 dr). Also in this case, the extended reaction time has no detrimental effect on the overall outcome. Using these conditions we were able to isolate diastereomerically pure major product 6 with reasonably good chemical yield of 87.5%. Diastereomers (S,2S)-6 and (S,2R)-7 were purified by column and fully characterized. The major product (S,2S)-6 gave [α]25D = +2616, indicating α-(S) configuration of the CF3-AA, while the minor diastereomer showed a negative sign of optical rotation ([α]25D = −1998), confirming α-(R) stereochemistry of the AA residue. The obtained data are in agreement of general trends in optical rotation observed for diastereomeric Ni(II)-complexes of this type [1f,5i,20].
As presented in Scheme 4, the disassembly of purified diastereomerically pure (>98% de) major product (S,2S)-6 was performed under the action of 3N aqueous HCl at 60 °C using dimethoxyethane (DME) as organic solvent. Virtually complete disappearance of complex (S,2S)-6 was observed within about 2 h of the reaction time. Upon cooling of the reaction mixture, the precipitate of salt of ligand (S)-5 was conveniently removed by filtration. The aqueous solution of the Ni(II) ions and free (S)-1, was concentrated, and treated with Fmoc-OSu in MeCN/H2O to provide the N-Fmoc protected amino acid (S)-9.
4. Conclusions
In summary, it was found that the application of new generation of chiral glycine equivalent (S)-4, prepared from commercially available ligand (S)-5, allows for convenient preparation of Fmoc-(S)-6,6,6-trifluoro-norleucine via alkylation reaction with CF3(CH2)3I. This protocol was consistently reproduced for synthesis of the target AA on ~10 g scale.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-8994/11/4/578/s1, Experimental procedures, full spectroscopic data for compounds 6, 7, and 9, and copies of 1H NMR, 13C, 19F NMR, and HPLC spectra (PDF).
Author Contributions
Conceptualization, H.M. and H.A.; Methodology, T.M.; Validation, H.M., Z.Y., and H.A.; Investigation, T.M.; Writing—Original Draft Preparation, J.H.; Writing—Review and Editing, V.A.S.; Supervision, H.M.
Funding
This research received no external funding.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21761132021) and IKERBASQUE, Basque Foundation for Science.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S. Unnatural amino acids in drug discovery. Chim. Oggi 2003, 21, 65–68. [Google Scholar]
- Hodgson, D.R.W.; Sanderson, J.M. The synthesis of peptides and proteins containing non-natural amino acids. Chem. Soc. Rev. 2004, 33, 422–430. [Google Scholar] [CrossRef]
- Sato, T.; Izawa, K.; Aceña, J.L.; Liu, H.; Soloshonok, V.A. Tailor-Made α-Amino Acids in the Pharmaceutical Industry: Synthetic Approaches to (1R, 2S)-1-Amino-2-vinylcyclopropane-1-carboxylic Acid (Vinyl-ACCA). Eur. J. Org. Chem. 2016, 2757–2774. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni (II) complexes of glycine Schiff bases; Part 1: Alkyl halide alkylations. Amino Acids 2013, 45, 691–718. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Izawa, K. Asymmetric Synthesis and Application of alpha-Amino Acids; ACS Symposium Series 1009; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α,α-Disubstituted-α-Amino Acids. Synthesis 2010, 14, 2319–2344. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluorine Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Smits, R.; Cadicamo, C.D.; Burger, K.; Koksch, B. Synthetic strategies to α-trifluoromethyl and α-difluoromethyl substituted α-amino acids. Chem. Soc. Rev. 2008, 37, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Kukhar, V.P.; Sorochinsky, A.E.; Soloshonok, V.A. Practical synthesis of fluorine containing alpha- and beta-amino acids: Recipes from Kiev, Ukraine. Future Med. Chem. 2009, 1, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of fluorine-containing amines, amino alcohols, α- and β-amino acids mediated by chiral sulfinyl group. J. Fluorine Chem. 2010, 131, 127–139. [Google Scholar] [CrossRef]
- Tarui, A.; Sato, K.; Omote, M.; Kumadaki, I.; Ando, A. Stereoselective synthesis of α-fluorinated amino acid derivatives. Adv. Synth. Catal. 2010, 352, 2733–2744. [Google Scholar] [CrossRef]
- Czekelius, C.; Tzschucke, C.C. Synthesis of halogenated carboxylic acids and amino acids. Synthesis 2010, 4, 543–566. [Google Scholar] [CrossRef]
- Qiu, X.-L.; Qing, F.-L. Recent advances in the synthesis of fluorinated amino acids. Eur. J. Org. Chem. 2011, 2011, 3261–3278. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Roeschenthaler, G.-V.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Recent Advances in the Asymmetric Synthesis of α-(Trifluoromethyl)-Containing α-Amino Acids. Synthesis 2012, 44, 1591–1602. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of fluorine containing α-amino acids in enantiomerically pure form via homologation of Ni(II) complexes of glycine and alanine Schiff bases. J. Fluorine Chem. 2013, 155, 21–38. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sánchez-Roselló, M.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Synthesis of fluorinated beta-amino acids. Synthesis 2011, 19, 3045–3079. [Google Scholar]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic Transamination—A Metal-Free Alternative to the Reductive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth. 2011, 8, 281–294. [Google Scholar] [CrossRef]
- Aceña, J.L.; Simon-Fuentes, A.; Santos, F. Recent Developments in the Synthesis of Fluorinated β-Amino Acids. Curr. Org. Chem. 2010, 14, 928–949. [Google Scholar] [CrossRef]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Urquhart, L. FDA new drug approvals in Q3 2018. Nat. Rev. Drug Discov. 2018, 17, 799. [Google Scholar]
- Scott, L.J. Eravacycline: A Review in Complicated Intra-Abdominal Infections. Drugs 2019, 79, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, T.; Kawada, K.; Ishihara, S.; Uchida, N.; Shiratori, O.; Higaki, J.; Hirata, M. Fluorine containing amino acids and their derivatives. 7. Synthesis and antitumor activity of α- and γ-substituted methotrexate analogs. Tetrahedron 1988, 44, 5375–5387. [Google Scholar] [CrossRef]
- Shu, M.; Yu, R.; Zhang, Y.; Wang, J.; Yang, L.; Wang, L.; Lin, Z. Predicting the activity of antimicrobial peptides with amino acid topological information. Med. Chem. 2013, 9, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I.; Jameison, F.A.; Pete, B.; Radunz, H.; Schittenhelm, C.; Lindner, H.J.; Emith, A.E. Design, synthesis and enzyme inhibitory activities of new trifluoromethyl-containing inhibitors for angiotensin converting enzyme. Drug Des. Discov. 1994, 11, 91–113. [Google Scholar] [PubMed]
- Borozan, S.Z.; Zlatović, M.V.; Stojanović, S.Đ. Anion–π interactions in complexes of proteins and halogen-containing amino acids. J. Biol. Inorg. Chem. 2016, 21, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- van Hest, J.C.M.; Kiick, K.L.; Tirrell, D.A. Efficient Incorporation of Unsaturated Methionine Analogues into Proteins in Vivo. J. Am. Chem. Soc. 2000, 122, 1282–1288. [Google Scholar] [CrossRef]
- Kiick, K.L.; Tirrell, D.A. Protein Engineering by In Vivo Incorporation of Non-Natural Amino Acids: Control of Incorporation of Methionine Analogues by Methionyl-tRNA Synthetase. Tetrahedron 2000, 56, 9487–9493. [Google Scholar] [CrossRef]
- Borozan, S.Z.; Stojanović, S.Đ. Halogen bonding in complexes of proteins and non-natural amino acids. Comput. Biol. Chem. 2013, 47, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, P.; Strandberg, E.; Heidenreich, N.; Bürck, J.; Fanghänel, S.; Ulrich, A.S. Self-Assembly of Flexible β-Strands into Immobile Amyloid-Like β-Sheets in Membranes As Revealed by Solid-State 19F NMR. J. Am. Chem. Soc. 2012, 134, 6512–6515. [Google Scholar] [CrossRef]
- Tkachenko, A.N.; Mykhailiuk, P.K.; Afonin, S.; Radchenko, D.S.; Kubyshkin, V.S.; Ulrich, A.S.; Komarov, I.V. 19F NMR Label to Substitute Polar Amino Acids in Peptides: A CF3-Substituted Analogue of Serine and Threonine. Angew. Chem. Int. Ed. 2013, 52, 1486–1489. [Google Scholar] [CrossRef]
- Gfeller, D.; Michielin, O.; Zoete, V. Expanding molecular modeling and design tools to non-natural sidechains. J. Comput. Chem. 2012, 33, 1525–1535. [Google Scholar] [CrossRef]
- Li, S.G.; Portela-Cubillo, F.; Zard, S.Z. A Convergent Synthesis of Enantiopure Open-Chain, Cyclic, and Fluorinated α-Amino Acids. Org. Lett. 2016, 18, 1888–1891. [Google Scholar] [CrossRef]
- Ojima, I.; Kato, K.; Nakahashi, K.; Fuchikami, T.; Fujita, M. New and effective routes to fluoro analogs of aliphatic and aromatic amino acids. J. Org. Chem. 1989, 54, 4511–4522. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Kukhar, V.P. Biomimetic Transamination of α-Keto Perfluorocarboxylic Esters. An Efficient Preparative Synthesis of β,β,β-Trifluoroalanine. Tetrahedron 1997, 53, 8307–8314. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Kirilenko, A.G.; Kukhar, V.P.; Resnati, G. Transamination of Fluorinated β-Keto Carboxylic Esters. A Biomimetic Approach to β-Polyfluoroalkyl-β-Amino Acids. Tetrahedron Lett. 1993, 34, 3621–3624. [Google Scholar] [CrossRef]
- Peng, W.; Wan, J.; Xie, B.; Ma, X. 9-Amino-(9-deoxy) cinchona alkaloid-derived new chiral phase-transfer catalysts. Org. Biomol. Chem. 2014, 12, 8336–8345. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.L.; Alsina, J.; Audu, C.O.; Babaev, E.; Cook, L.; Dage, J.L.; Goodwin, L.A.; Martynow, J.G.; Matosiuk, D.; Royo, M.; et al. Distributed Drug Discovery, Part 2: Global Rehearsal of Alkylating Agents for the Synthesis of Resin-Bound Unnatural Amino Acids and Virtual D3 Catalog Construction. J. Comb. Chem. 2009, 11, 14–33. [Google Scholar] [CrossRef]
- Yajima, T.; Nagano, H. Photoinduced Diastereoselective Addition of Perfluoroalkyl Iodides to Acrylic Acid Derivatives for the Synthesis of Fluorinated Amino Acids. Org. Lett. 2007, 9, 2513–2515. [Google Scholar] [CrossRef]
- Larsson, U.; Carlson, R.; Leroy, J. Synthesis of amino acids with modified principal properties 1. Amino acids with fluorinated side chains. Acta Chem. Scand. 1993, 47, 380–390. [Google Scholar] [CrossRef]
- Wang, J.; Lin, D.; Zhou, S.; Soloshonok, V.A.; Liu, H. Asymmetric Synthesis of Sterically Constrained Linear Trifluoromethyl Containing Amino Acids via Alkylation of Chiral Equivalents of Nucleophilic Glycine and Alanine. J. Org. Chem. 2011, 76, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Gerus, I.I.; Yagupolskii, Y.L.; Kukhar, V.P. Fluorine-Containing Amino Acids. III. α-Trifluoromethyl-α-Amino Acids. Zh. Org. Khim. 1987, 23, 2308–2313. [Google Scholar]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally Convenient Asymmetric Synthesis of (S)- and (R)-3-Amino-4,4,4-trifluorobutanoic Acid. Part II: Enantioselective Biomimetic Transamination of 4,4,4-Trifluoro-3-oxo-N-[(R)-1-phenylethyl)butanamide. J. Fluorine Chem. 2006, 127, 930–935. [Google Scholar] [CrossRef]
- Röschenthaler, G.-V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Poliashko, K.O.; Kukhar, V.P.; Rozhenko, A.B.; Soloshonok, V.A.; Sorochinsky, A.E. Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun. 2012, 48, 11519–11521. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J. A Practical Asymmetric Synthesis of Enantiomerically Pure 3-Substituted Pyroglutamic Acids and Related Compounds. Angew. Chem. Int. Ed. 2000, 39, 2172–2175. [Google Scholar] [CrossRef]
- Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V.A. Efficient Asymmetric Synthesis of Novel 4-Substituted and Configurationally Stable Analogs of Thalidomide. Org. Lett. 2006, 8, 5625–5628. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019. [Google Scholar] [CrossRef] [PubMed]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 2: Aldol, Mannich addition reactions, deracemization and (S) to (R) interconversion of α-amino acids. Amino Acids 2013, 45, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Recent approaches for asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes. Amino Acids 2017, 49, 1487–1520. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V.A. The Design, Synthesis and Evaluation of a New Generation of Modular Nucleophilic Glycine Equivalents for the Efficient Synthesis of Sterically Constrained α-Amino Acids. J. Org. Chem. 2006, 71, 8572–8578. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H.; Ellis, T.K.; Yamada, T.; Ohfune, Y. Application of Modular Nucleophilic Glycine Equivalents for Truly Practical Asymmetric Synthesis of β-Substituted Pyroglutamic Acids. Tetrahedron Lett. 2005, 46, 1107–1110. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ellis, T.K.; Ueki, H.; Ono, T. Resolution/Deracemization of Chiral α-Amino Acids Using Resolving Reagents with Flexible Stereogenic Centers. J. Am. Chem. Soc. 2009, 131, 7208–7209. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J.L.; et al. Chemical Dynamic Kinetic Resolution and (S)/(R)-Interconversion of Unprotected α-Amino Acids. Angew. Chem. Int. Ed. 2014, 53, 12214–12217. [Google Scholar] [CrossRef]
- Bergagnini, M.; Fukushi, K.; Han, J.; Shibata, N.; Roussel, C.; Ellis, T.K.; Aceña, J.L.; Soloshonok, V.A. NH-type of chiral Ni(II) complexes of glycine Schiff base: Design, structural evaluation, reactivity and synthetic applications. Org. Biomol. Chem. 2014, 12, 1278–1291. [Google Scholar] [CrossRef]
- Nian, Y.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Analysis of crystallographic structures of Ni(II) complexes of α-amino acid Schiff bases; Elucidation of the substituents effect on stereochemical preferences. Dalton Trans. 2017, 46, 4191–4198. [Google Scholar] [CrossRef]
- Tang, X.; Soloshonok, V.A.; Hruby, V.J. Convenient Asymmetric Synthesis of Enantiomerically Pure 2′,6′-Dimethyltyrosine (DMT) via Alkylation of Chiral Nucleophilic Glycine Equivalent. Tetrahedron Asymmetry 2000, 11, 2917–2925. [Google Scholar] [CrossRef]
- Nian, Y.; Wang, J.; Zhou, S.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V.A.; Liu, H. Recyclable Ligands for the Non-Enzymatic Dynamic Kinetic Resolution of Challenging α-Amino Acids. Angew. Chem. Int. Ed. 2015, 54, 12918–12922. [Google Scholar] [CrossRef] [PubMed]
- Nian, Y.; Wang, J.; Zhou, S.; Dai, W.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V.A.; Liu, H. Purely Chemical Approach for Preparation of D-alpha-amino Acids via (S)-to-(R)-interconversion of Unprotected Tailor-made alpha-amino Acids. J. Org. Chem. 2016, 81, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Chen, X.; Aceña, J.L.; Soloshonok, V.A.; Liu, H. Chemical Kinetic Resolution of Unprotected β-Substituted-β-Amino Acids Using Recyclable Chiral Ligands. Angew. Chem. Int. Ed. 2014, 53, 7883–7886. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Hiramatsu, T.; Takeda, R.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Expedient Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid via Alkylation of Chiral Nucleophilic Glycine Equivalent. Org. Process. Res. Dev. 2019. [Google Scholar] [CrossRef]
- Romoff, T.T.; Palmer, A.B.; Mansour, N.; Creighton, C.J.; Miwa, T.; Ejima, Y.; Moriwaki, H.; Soloshonok, V.A. Scale-up Synthesis of (R)- and (S)-N-(2-benzoyl-4-chlorophenyl)-1-(3,4-dichlorobenzyl)pyrrolidine-2-carboxamide hydrochloride, a Versatile Reagent for Preparation of Tailor-made α- and β-Amino Acids in Enantiomerically Pure Form. Org. Process. Res. Dev. 2017, 21, 732–739. [Google Scholar] [CrossRef]
- Ueki, H.; Ellis, T.K.; Martin, C.H.; Soloshonok, V.A. Efficient Large-Scale Synthesis of Picolinic Acid Derived Ni(II)-Complexes of Glycine. Eur. J. Org. Chem. 2003, 2003, 1954–1957. [Google Scholar] [CrossRef]
- Ellis, T.K.; Hochla, V.M.; Soloshonok, V.A. Efficient Synthesis of 2-Aminoindane-2-Carboxylic Acid via Dialkylation of Nucleophilic Glycine Equivalent. J. Org. Chem. 2003, 68, 4973–4976. [Google Scholar] [CrossRef] [PubMed]
- Houck, D.; Aceña, J.L.; Soloshonok, V.A. Alkylations of Chiral Nickel(II) Complexes of Glycine under Phase-Transfer Conditions. Helv. Chim. Acta 2012, 95, 2672–2679. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H. Design, Synthesis and Characterization of Binuclear Ni(II) Complexes with Inherent Helical Chirality. J. Am. Chem. Soc. 2007, 129, 2426–2427. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ono, T.; Ueki, H.; Vanthuyne, N.; Balaban, T.S.; Bürck, J.; Fliegl, H.; Klopper, W.; Naubron, J.V.; Tam, T.T.; et al. Ridge-tile-like chiral topology: Synthesis, resolution and complete chiroptical characterization of enantiomers of edge-sharing binuclear square planar complexes of Ni(II) bearing achiral ligands. J. Am. Chem. Soc. 2010, 132, 10477–10483. [Google Scholar] [CrossRef]
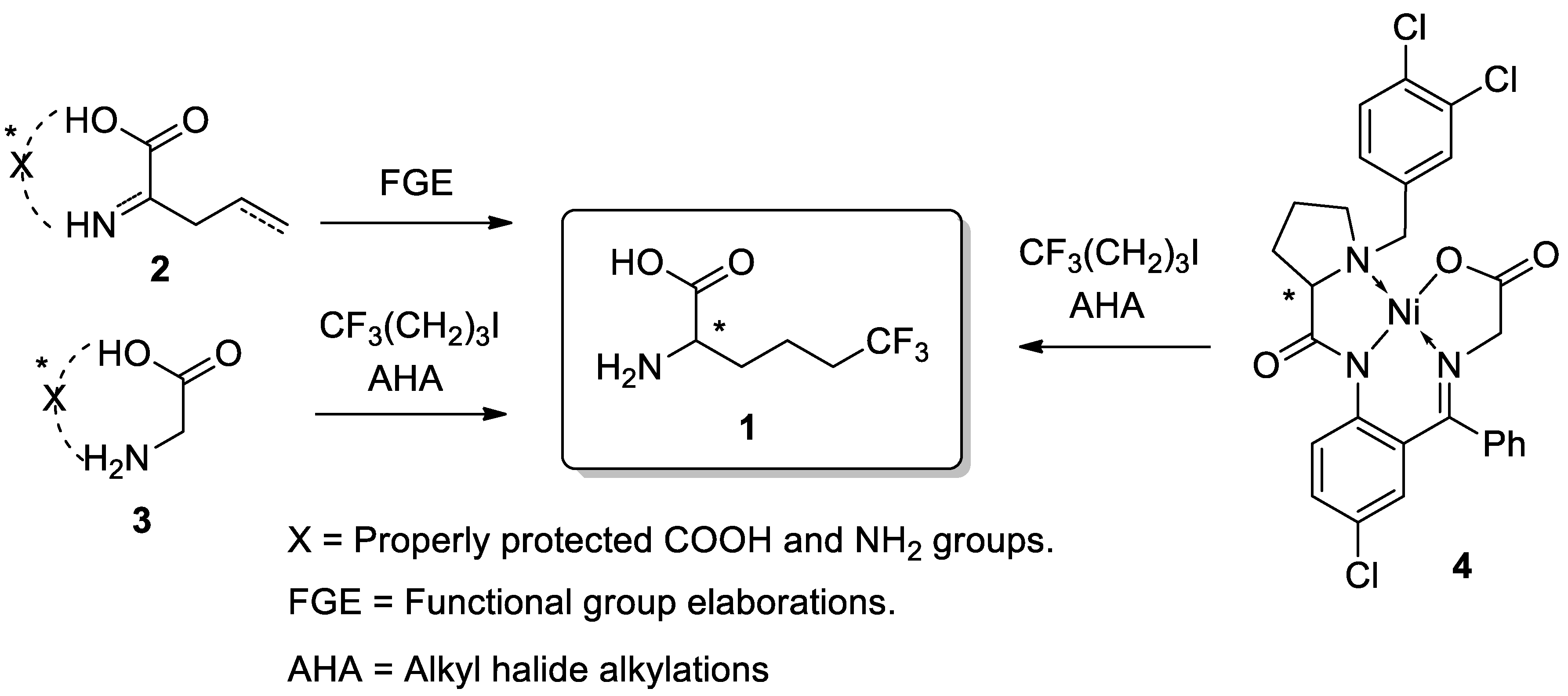
Scheme 1.
Literature methods for synthesis of 6,6,6-trifluoro-norleucine (1) via functional group elaborations (FGE) in 2 and alkyl halide alkylations (AHA) of glycine derivatives (3). Application of chiral Ni(II) complexes (4) for preparation of 1 via AHA.
Scheme 1.
Literature methods for synthesis of 6,6,6-trifluoro-norleucine (1) via functional group elaborations (FGE) in 2 and alkyl halide alkylations (AHA) of glycine derivatives (3). Application of chiral Ni(II) complexes (4) for preparation of 1 via AHA.

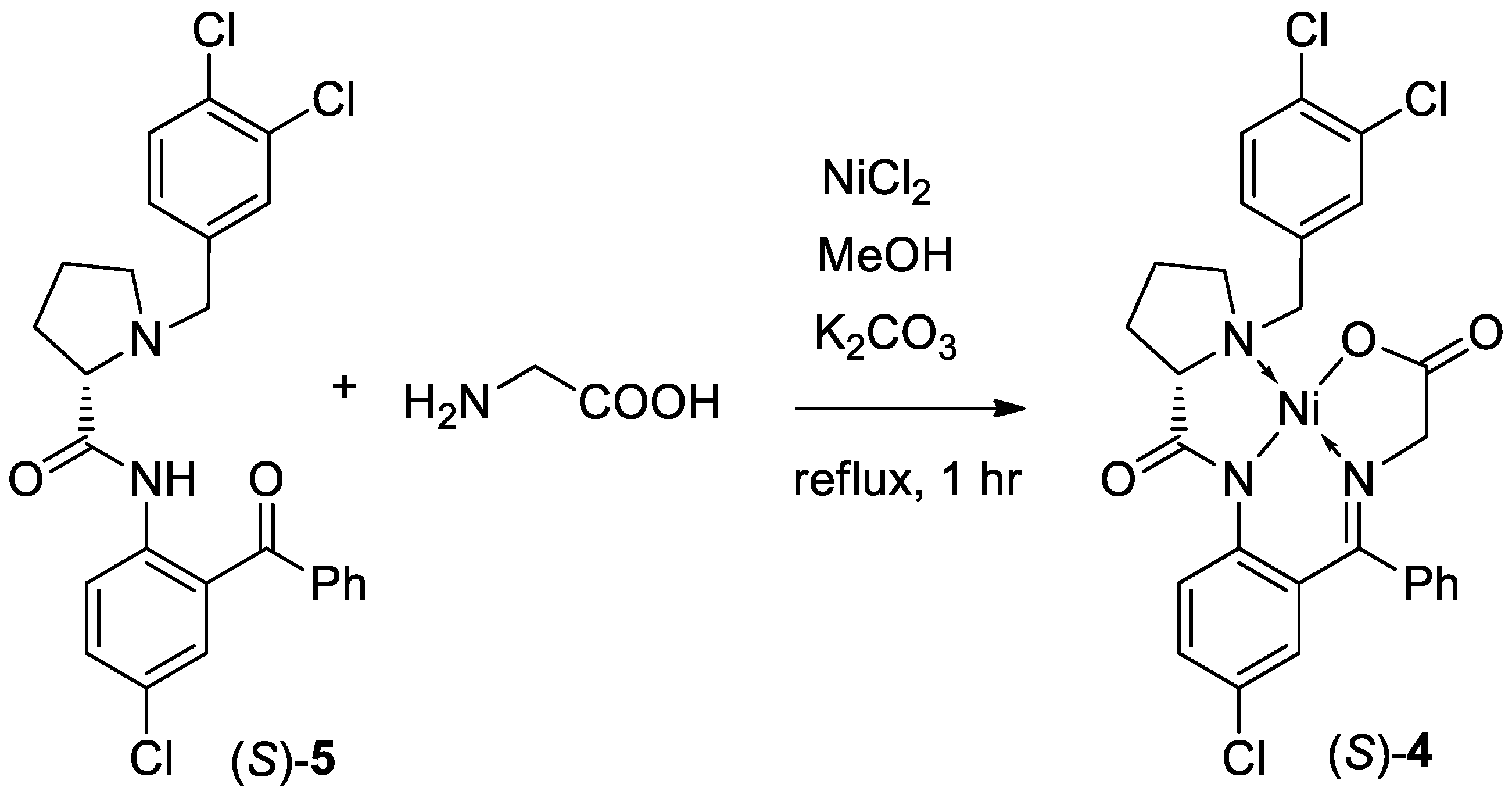
Scheme 2.
Synthesis of chiral Ni(II) complex of glycine Schiff base 4.

Scheme 3.
Alkylation of (S)-3 with CF3(CH2)3I under homogeneous conditions.

Scheme 4.
Disassembly of diastereomerically pure (S,2S)-6, recovery of chiral ligand (S)-5, and isolation of Fmoc-(S)-6,6,6-trifluoro-norleucine 9.
Scheme 4.
Disassembly of diastereomerically pure (S,2S)-6, recovery of chiral ligand (S)-5, and isolation of Fmoc-(S)-6,6,6-trifluoro-norleucine 9.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Reaction of complex (S)-4 with CF3(CH2)3I in DMSO using solid NaOH as a base [a].
| Compounds | Byproducts | ||||||
|---|---|---|---|---|---|---|---|
| Entry | Time (h) | 4 (%) | 6 (%) | 7 (%) | Dr (6:7) | 5 (%) | 8 + uk [b] (%) |
| 1 | 1.5 | 63.9 | 21.7 | 4.0 | 84:16 | 0.7 | 3.0 |
| 2 | 3.0 | 54.5 | 25.7 | 4.7 | 85:15 | 0.4 | 5.1 |
| 3 | 4.5 | 43.6 | 27.8 | 5.3 | 84:16 | 0.6 | 8.9 |
| 4 | 24.0 | 0.7 | 37.9 | 2.1 | 95:5 | 5.1 | 42.5 |
[a] Reaction conditions: complex (S)-4, DMSO (20 v/w), NaOH (1.0 equiv), CF3(CH2)I (1.0 equiv). [b] unknown compounds.
Table 2.
Reaction of complex (S)-4 with CF3(CH2)3I in DMF using solid NaOMe (28% and 10% solution in MeOH) as a base [a].
Table 2.
Reaction of complex (S)-4 with CF3(CH2)3I in DMF using solid NaOMe (28% and 10% solution in MeOH) as a base [a].
| Compounds | Byproducts | |||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Time (h) | NaOMe (Concentration) | 4 (%) | 6 (%) | 7 (%) | Dr (6:7) | 5 (%) | 8 + uk [b] (%) |
| 1 | 0.5 | 28% | 0.9 | 81.3 | 9.0 | 90:10 | 1.15 | 0.2 |
| 2 | 2.0 | 28% | <0.1 | 82.1 | 9.1 | 90:10 | 1.8 | 0.35 |
| 3 | 0.5 | 10% | 0.2 | 89.05 | 3.2 | 97:3 | 0.3 | 2.5 |
| 4 | 2.0 | 10% | 0.2 | 89.3 | 3.25 | 96.5:3.5 | 0.35 | 3.7 |
[a] Reaction conditions: complex (S)-4, DMF (7 v/w), NaOMe/MeOH (1.0 equiv), CF3(CH2)I (1.0 equiv). Isolated yield of 6 was 87.5%. [b] unknown compounds.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine. Symmetry 2019, 11, 578. https://0-doi-org.brum.beds.ac.uk/10.3390/sym11040578
AMA Style
Mei H, Yin Z, Miwa T, Moriwaki H, Abe H, Han J, Soloshonok VA. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine. Symmetry. 2019; 11(4):578. https://0-doi-org.brum.beds.ac.uk/10.3390/sym11040578
Chicago/Turabian StyleMei, Haibo, Zizhen Yin, Toshio Miwa, Hiroki Moriwaki, Hidenori Abe, Jianlin Han, and Vadim A. Soloshonok. 2019. "Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine" Symmetry 11, no. 4: 578. https://0-doi-org.brum.beds.ac.uk/10.3390/sym11040578
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.