1. Introduction
The origin of homochirality remains one of the profound mysteries of life’s evolution. Why do all living things on Earth utilize L-amino acids and D-sugars? Hypotheses on this subject date back to the discovery of homochirality by Pasteur and can be divided into those that propound the existence of an asymmetric force which selects for preferred chirality [
1,
2,
3] and those who propose it arose through a “frozen accident”. The difficulty with “frozen accident” theories is that they can never be tested. Moreover, at present, there is no experimental evidence demonstrating that any putative asymmetric force produces preferred chirality, and such evidence would create a conundrum of which advocates generally seem to be unaware: any asymmetric force would work equally on organic and inorganic molecules, and within living and non-living systems. Thus, evidence for an asymmetric force would create a new problem, which would be why that force is manifested in living systems exclusively. I have therefore spent many years [
4] attempting to rethink the problem of homochirality and have recently [
5] recognized an assumption (usually, though not always, unstated) that homochirality emerged before the code itself. But what if homochirality was
not established before the genetic code emerged? I began exploring the implications of this unexplored possibility and two years ago proposed a radically different hypothesis: the reason that homochirality and an essentially universal genetic code both characterize living organisms may be that they arose as a function of each other [
5].
Like much other work on the origin of the genetic code, this co-evolution of the code and homocharility (COCAH) hypothesis makes the testable assumption that the genetic code arose through amino acid-codon pairing. Several investigators have provided significant experimental evidence for amino acids binding to their codons [
6,
7,
8,
9,
10,
11,
12,
13,
14], but with only two exceptions these experiments have been performed using L-amino acids and D-ribonucleotides. The two exceptions were Yarus [
11] and Profy and Uster [
15], who demonstrated that D-codons prefer an L-amino acid to a D-amino acid. Indeed, theoretical calculations by Han,
et al. [
16] suggest the more general conclusion that D-ribonucleotides should preferentially select the L-amino acids over their D-counterparts. By extrapolation, L-ribonucleotides should preferentially select D-amino acids. No one, however, has experimentally tested the binding of D-amino acids to L-codons.
A second assumption that is implicit in the literature on binding of amino acids to their codons is that this binding is directionally sensitive. That is to say, if an amino acid binds preferentially to the sequence 5’-ACG-3’, then it will bind to the inverse sequence, 5’-GCA-3’, with lesser affinity. If that were not true, then the codons ACG and GCA would encode the same amino acids rather than different ones. I, however, have suggested previously that during the evolution of the genetic code, translation direction may have been ambiguous [
4,
5]. Specificity of amino acid binding to codons of opposite directionality therefore becomes a problem in need of experimental testing. If the binding is insensitive to directionality, then the origin of the direction in which the code is “read” is also a problem that may have been solved during the origin of the code itself.
The COCAH co-evolution hypothesis [
5] proposes that the genetic code, its translation direction, and homochirality emerged through a common natural selection process. Like previous work in this area (see above) the COCAH hypothesis requires that chiral preferences be expressed in amino acid binding to codons: L-amino acids should prefer D-codons and D-amino acids, L-codons. The hypothesis also makes a number of unique, testable predictions. First, the code was directionally ambiguous, so that every codon had at least two different amino acid assignments (one read 5’>3’ and the other, 3’>5’). Second, multiple codes therefore evolved in which different amino acids competed for the same codons. Third, selection for an unambiguous set of codon assignments was determined by a combination of binding affinity, amino acid prevalence, and codon prevalence, which resulted in a preference for L- amino acids pairing with D-codons. Fourth, the direction of the encodings was also determined by this selection process. The present research experimentally tests the first three of these predictions and makes testable predictions concerning what conditions would have had to exist to result in the current set of genetic encodings, code directionality, and homochirality. The hypothesis also suggests that under different prebiotic conditions, a different code with a different set of preferred homochiralities could have emerged.
Binding studies provide the basis of the tests of the COCAH coevolution-of-code-and-homochirality hypothesis. According to the modern genetic code (read 5’→3’), CGU codes for arginine (Arg) and its inverse codon, UGC encodes cysteine (Cys). Similarly, GUA encodes valine (Val) while its inverse, AUG, encodes methionine (Met). A tetramer CGUA would therefore encode both Arg (in the first three bases) and Val (in the second three), while AUGC would encode both Met and Cys. If the original genetic code were a doublet (as some investigators have hypothesized), then the tetramer CGUA would encode three amino acids: CG = Arg, GU = Val, and UA = Tyrosine (Tyr). Similarly, AUGC would also encode three amino acids: AU = Met, UG = Cys, and GC = Alanine (Ala). On the other hand, if the code were ambiguous in terms of either direction or chiral specificity or both, then the modern encodings may disguise a more complicated set of amino acid-codon interactions. By comparing the binding affinities of various L- and D-amino acids under identical conditions to both L- and D-oligonucleotides composed of identical bases and to their ordered inverses, it should be possible to determine whether amino acids have specificity for base sequence, codon direction, codon length (triplet or doublet), and codon chirality. The tests carried out below are first time this set of factors has been investigated simultaneously.
2. Results and Discussion
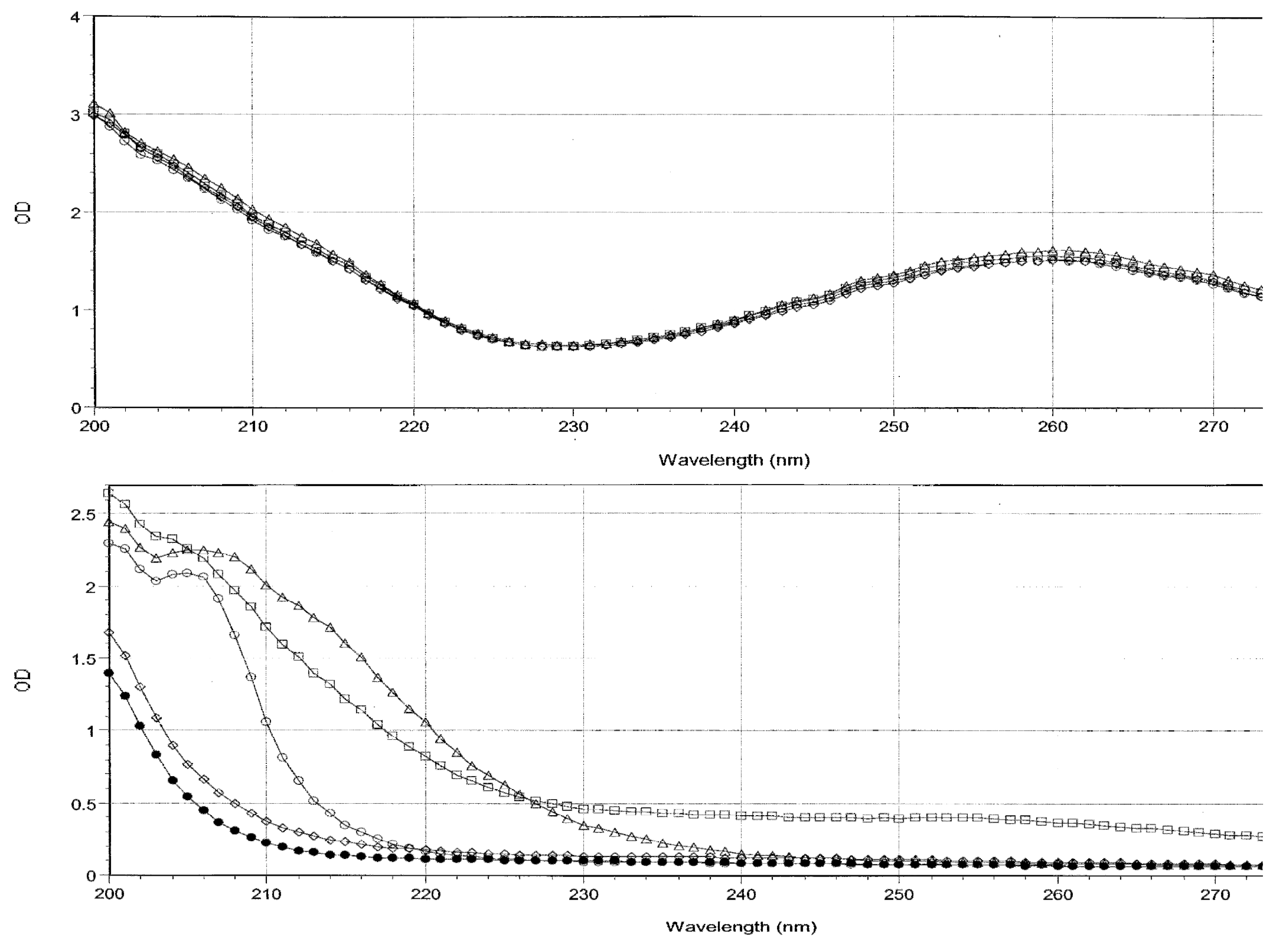
Spectra of the starting solutions of the four oligonucleotides and of the amino acids were obtained (
Figure 1). These spectra demonstrate that the optical density of the four oligonucleotide solutions was almost exactly identical so that no significant differences in binding to amino acids can be attributed to differences in oligonucleotide concentration.
The majority of spectral absorption occurs in the range between 200 and 220 nm for both the oligonucleotides and the peptides after subtracting out the buffer contribution. Data from experiments combining the oligonucleotides with amino acids were analyzed at the 200, 205, 210, 215, 225, 235, and 260 nm regions for combinations of Arg, Met, Cys and Val with each of the oligonucleotides (data not shown) to determine the spectral range in which binding would be most visible. Measurable interactions were observable at 200, 205, and 210 nm, but dropped off significantly as the wavelength increased, and beyond 220 nm, binding became difficult or impossible to detect. Such differences are not surprising, as many other studies have shown that the amount of change in absorbance that results from the binding of two molecules differs at different wavelengths depending on which residues or components of the molecules are involved in binding (see, for example, Figure 4 of Nafisi,
et al. [
17]; Figure 2 of Giri and Kumar [
18]; and Dillon,
et al., [
19]).
Notably, the relative binding constants determined at 200 nm tend to be about ten times greater than those measured at 210 nm suggesting that specific molecular constituents of the oligonucleotides and amino acids that absorb at 200 are more involved in binding than are constituents that absorb at 210 or higher wavelengths. Since absorbances close to 190 usually involve molecular interactions with water, these data suggest that hydrogen and ionic bonds are the key players in the interactions reported here, as opposed to changes in base stacking, intercalation and charge transfer complexation, which tends to show up in absorbances near 260 nm [
20,
21]. Significantly, there was no binding observable at all for any amino acid in the 260 range, possibly because none of the amino acids had any observable absorbance at this wavelength and they are very unlikely to have intercalated into the ring structure of the oligonucleotides, which is what creates the majority of oligonucleotide absorbance at this wavelength. The lack of changes at 260 may therefore suggests that the stacking of the bases in the oligonucleotides is not affected by amino acid binding. As a result of these experiments, data analysis was limited to the 200 and 210 wavelengths for the rest of the amino acids tested (Gly, Ala, Ser, Thr, Asp, Glu, Leu, Pro, Phe).
Because the binding constants calculated at 200 nm and 210 nm wavelengths differ, it is impossible to say with certainty what the absolute affinity for any given amino acid is for any given oligonucleotide (which is why calculations involving interacting molecules studied using UV spectroscopy always specify the wavelength from which the binding constant was calculated). Importantly, no significant differences in the
ratios of binding constants at different wavelengths were observed (
Table 1). Since the hypothesis being tested here depends not on the absolute affinities, but on the relative affinities of each amino acid for each oligonucleotide at under the same conditions, it does not appear to matter at what wavelength binding constants are calculated as long as observable binding can be measured.
All of the amino acids (Arg, Met, Cys and Val) encoded in the modern genetic code by the sequences CGUA and AUCG bind with measurable affinities (most in the micromolar range) to these oligonucleotides, while most of the other nine amino acids have relatively small (millimolar) or unmeasurable affinities (
Table 1).
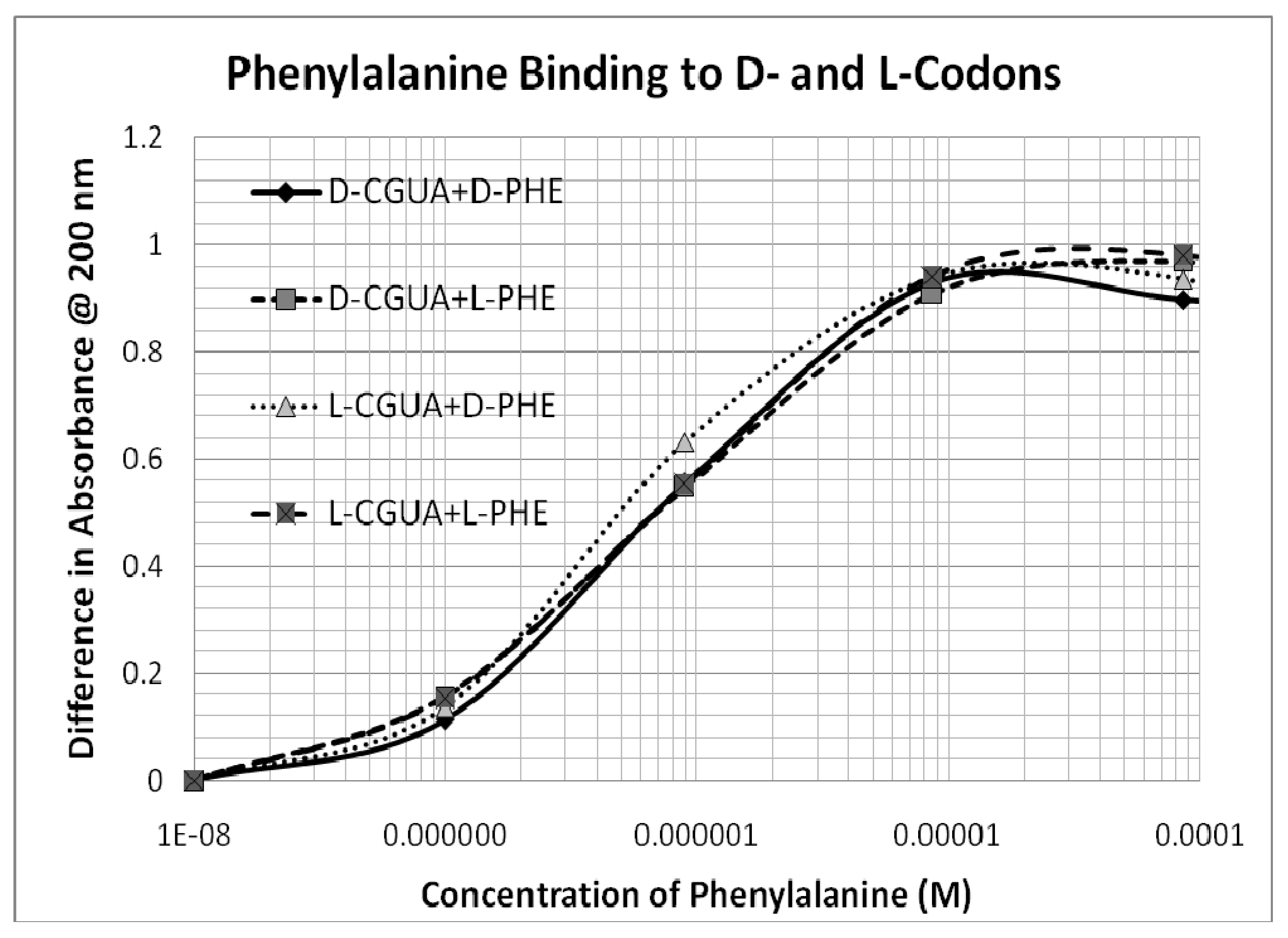
The one major exception is Phe, which binds with micromolar affinity to all four oligonucleotides (
Table 1 and
Figure 2). Unlike the encoded amino acids, which will be discussed in greater detail below, Phe binding to the oligonucleotides is not affected by the chirality of either the amino acid or that of the oligonucleotides and may be due to non-specific intercalation into the bases.
Several other cases of chiral- and direction-insensitive binding were also observed, including Gly (
Figure 3a), Pro (
Figure 3b), Thr, Leu, and Ala (spectra not shown; see binding constants in
Table 1).
The cases of chirally insensitive and direction-insensitive binding suggest that the precision of the technique used to determine binding constants is quite good: variance in measured binding constants among the various combinations of these non-coded amino acid and the oligonucleotides is generally within 20 to 40%. In contrast, the differences in binding constants measured among the various combinations of amino acids encoded by the oligonucleotides was in the range of two- to three-fold (200–300%,
i.e., an order of magnitude greater) (
Table 1 and
Figure 4,
Figure 5,
Figure 6 and
Figure 7).
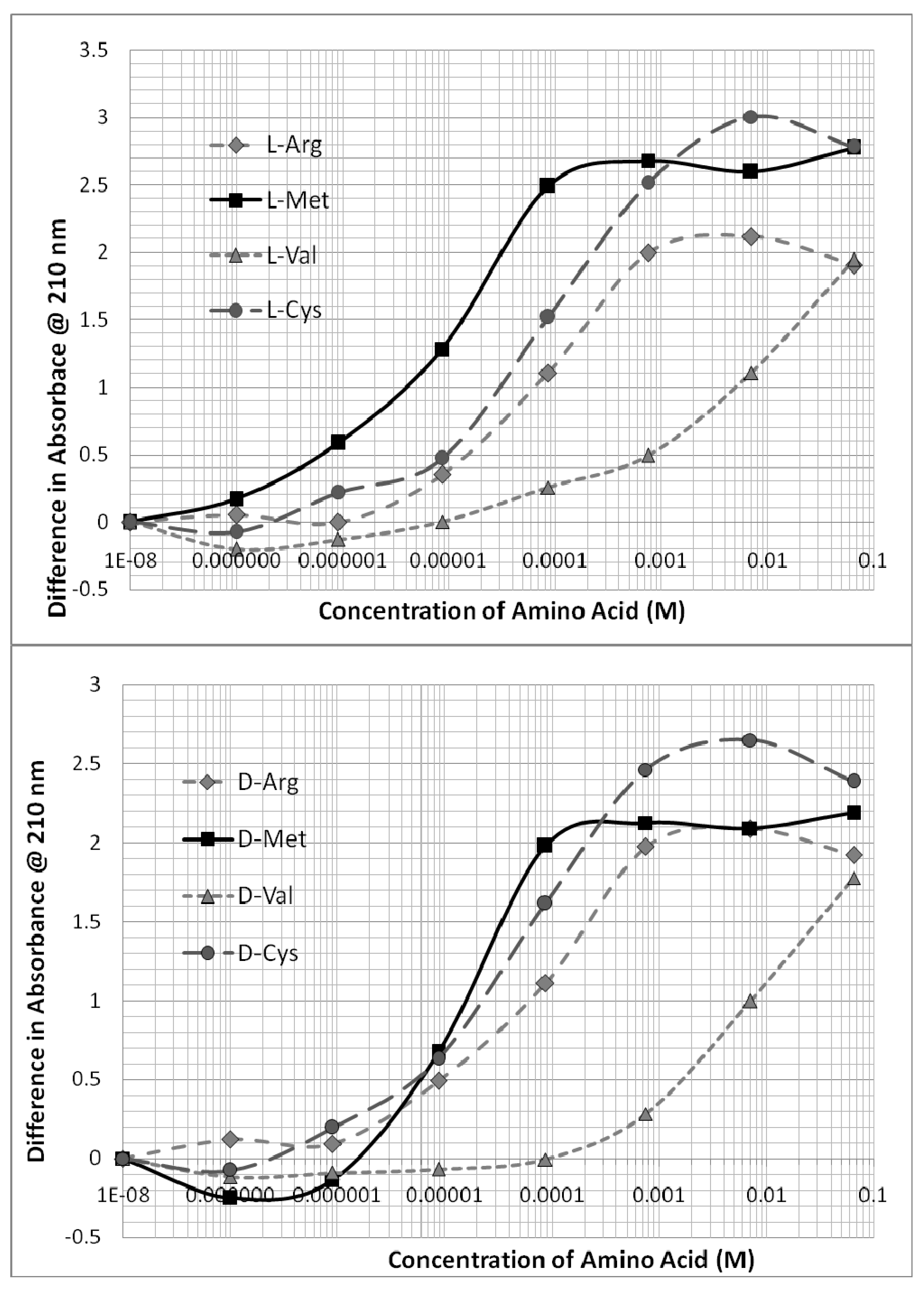
Figure 4 demonstrates that the technique used is able to distinguish clearly between the binding of different amino acids of the same chirality to any given oligonucleotide. Arg, Met, Cys, and Val each bind to the oligonucleotides that include their modern genetic encodings. The binding of any given chiral form of these amino acids is weakly sensitive to both the chirality and direction of the oligonucleotide such that the resulting binding constants vary accordingly (
Table 1). In general, L-amino acids prefer D-oligonucleotides, and D-amino acids prefer L-oligonucleotides, but there is one exception, which is that both L- and D-Cys consistently preferred D-oligonucleotides (
Figure 4 and
Figure 5). This preference is observable by comparing the two portions of
Figure 4 with each other.
Figure 5 presents data showing that the D-forms of L- and D-Arg and Cys have significantly greater affinity for L-AUGC than do the D- amino acids (compare with
Figure 2 and
Figure 3).
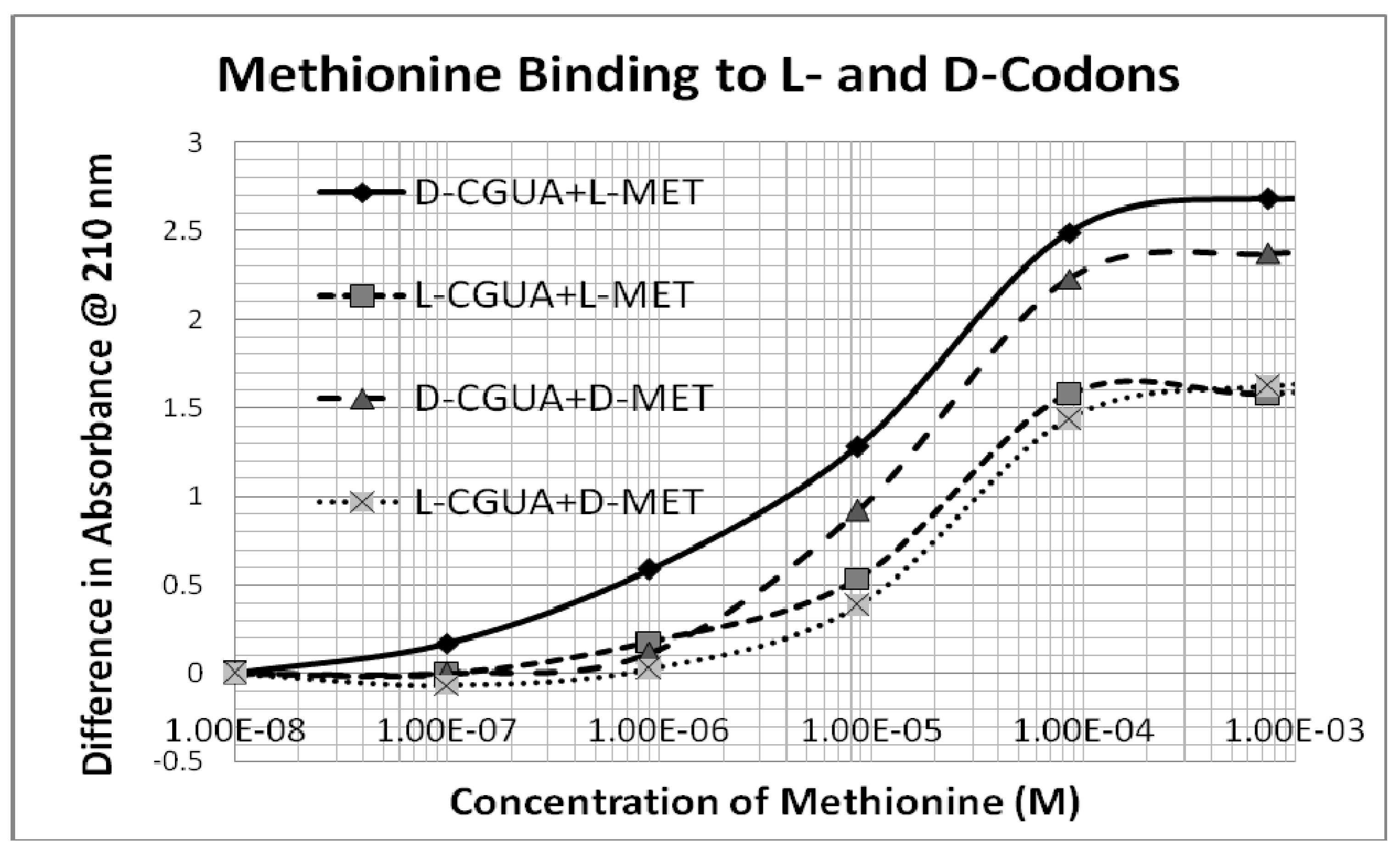
Figure 6 presents typical data showing several-fold stronger binding of L-Met to D-CGUA than other Met-CGUA combinations.
Figure 7 presents similar data showing similar several-fold differences in the binding of D-Arg to the four oligonucleotides. These and their related data, as summarized in
Table 1, provide evidence that the binding of encoded amino acids to their encoding oligonucleotides appears to be sensitive to both chirality and to direction of encoding within a two- to three-fold range of affinities. To repeat an important point made above, the binding of non-encoded amino acids (e.g., Gly, Pro, Leu, Thr, Ala) to these same oligonucleotides is, in general, less strong than the encoded amino acids, and displays relatively less sensitivity to chirality and directionality (
Table 1 and
Figure 2 and
Figure 3).
In sum, two D-RNA-oligonucleotides having inverse base sequences (D-CGUA and D-AUGC) and their corresponding L-RNA-oligonucleotides (L-CGUA and L-AUGC) were synthesized and compared with regard to their ability to bind L- and D-amino acids. According to the modern genetic code, these oligonucleotides (read according to the modern convention of 5’→3’) encode arginine (CGU), valine (GUA), methionine (AUG) and cysteine (UGC). The resulting binding constants demonstrate that: (1) all tested oligonucleotides bound multiple encoded amino acids but with different affinities; 2) encoded amino acids bind in the micromolar range (the Kd at 200 nm ranged from 130 to 500 nM for Arg; 750 nM to 6 uM for Met; 3 to 80 uM for Cys; and 370 to 900 uM for Val); (2) there is in general a two- to three-fold preference of encoded D-amino acids for L-codons and L-amino acids for D-codons; (3) encoded amino acids show a two- to three-fold preference for their appropriate codon sequence; (4) with the notable exception of Phe, the oligonucleotides had very low affinity for non-encoded amino acids; (5) most non-coded amino acids showed about a ten-fold reduced preference for oligonucleotide sequence or chirality compared with encoded amino acids.
4. Conclusions
The data presented in
Table 1 shows that Arg, Met, Cys and Val bind to CGUA and AUGC as predicted by the modern genetic code. These data therefore support the hypothesis that the genetic code emerged as a result of selection by direct binding between amino acids and their codons [
9] and are consistent with previous experiments showing that other amino acids bind to their codons [
6,
7,
8,
9,
10,
11,
12,
13,
14]. No significant binding was observed between L- or D-Ala and CGUA or AUGC codons, as might have been expected if the original code was doublet in nature, so these data support a triplet origin for the code.
There is a two- to three-fold selection for chirality. L-amino acids generally prefer D-codons (with the notable exception of D-cysteine, which prefers D-CGUA, a point of some interest that will be discussed below). There is also some selectivity for codon directionality as well, but this is usually not more than two-fold. Thus, L-amino acids often have more similar affinities for both D-CGUA and D-AUGC, or for both L-CGUA and L-AUGC than for the pair D-CGUA and L-CGUA, or for the pair L-AUGC and D-AUGC. Both of these findings are consistent with the expectations of the COHAC theory.
The differences within the binding affinities of each encoded amino acid for the different oligonucleotides is about an order of magnitude greater than the differences observed in the binding of each non-encoded amino acid for the same oligonucleotides, suggesting that the encoded amino acids are more sensitive to chirality and direction of the oligonucleotides than non-encoded ones. The simplest explanation of this observation is that there is both specific and non-specific interaction involved in amino acid-codon binding. The non-specific interaction may involve simple hydrogen or ionic-bonding between amino and carboxyl groups of the amino acids to the nucleosides. The specific interactions presumably involve side chain interactions as well, which involve chiral stereochemical bonding. The method used here is unable to test this conjecture, which might be explored more fully with nuclear magnetic resonance.
In addition, the encoded amino acids (with the exception of Val) bind several orders of magnitude more tightly to their encoding oligonucleotides than do the non-encoded amino acids (with the notable exception of Phe, the binding of which appears to be non-specific). Thus, there is a reasonable, if not complete, correlation between the affinities observed here and the structure of the modern genetic code. The exceptions will be discussed below.
These data suggest that origin of the genetic code could have occurred under racemic conditions that would have led to chiral- and direction-specific interactions between amino acids and their codons. In general, L-amino acids would have preferred D-codons, and D-amino acids, L-codons. The amino acids would have bound to both the codon sequence and its inverse with only a two- or three-fold difference in affinity. Thus, every codon would encode multiple amino acids and both chiral forms of these amino acids (as demonstrated here) with only small differences. Multiple, competing genetic codes would have been likely. In order to understand how these multiple codes may have yielded the one code that we observe today, and the specific forms of homochirality that characterize amino acids and nucleic acids today, it is necessary to put these results in a wider context.
A very important assumption informing this research is that prior to the origin of the genetic code, homochirality had not been established in any chemical species on Earth. This assumption does not, however, preclude processes by which selection for
racemic mixtures of homopolymers of L- or D-constituents were produced. For example, previous experiments by Orgel [
26] and Eschenmoser [
27] have demonstrated that RNA ligation and template-directed synthesis are chirally-sensitive so that chiral mixtures of RNA monomers will produce racemic mixtures of homochiral polymers made up of exclusively L- or exclusively D-ribonucleotides. We can therefore assume with some assurance that prior to and during the emergence of the genetic code, the precursors of codons and anticodons were racemic mixtures of homochiral oligonucleotides rather than polymers composed of disordered or random L- and D-constituents. The preferential emergence of racemic mixtures of chirally pure L- and D-RNA codons (or anticodons) set the stage for the emergence of the genetic code. It is likely that the same processes of ligation combined with template-directed synthesis of complementary polyribonucleotides selected among all of the possible bases that may have been available prebiotically. In consequence, only a few, including adenine, uracil, cytosine, and guanine were preferentially incorporated into these racemic mixtures of homochiral RNAs.
A similar process of selection probably narrowed the range of available amino acids [
28]. On the one hand, it is well established that certain amino acids are more likely than others to be produced under conditions modeling prebiotic chemistries, and these include glycine, alanine, proline, valine, leucine, and arginine, whereas structurally complex or difficult to synthesize compounds such as phenylalanine, tyrosine, and tryptophan may have been very rare [
29,
30,
31,
32,
33,
34]. Of equal importance to synthesis of diverse amino acids, however, would have been means for selective retention of some subset of them. Highly reactive compounds such as cysteine and methionine, for example, could very well have been synthesized in reasonable amounts (especially in sulphur-rich prebiotic conditions) but would have rapidly disappeared [
35,
36] if not stabilized by non-covalent complexing to compounds such as oligomers, as we have demonstrated here. Two forms of amino acid complementarity may have provided selection for the basic twenty amino acids incorporated into living systems. Grafstein [
37] has demonstrated that only a very select subset of possible amino acids can form very stable cyclical dipeptides (diketopiperizines) in which the side chains of the amino acids (one L- and one D-) are able to form stereochemically specific complexes across the molecule’s ring structure. These amino acids are the twenty used by modern living systems. Incorporation of select amino acids into diketopiperazines would therefore have resulted in retention of these, while amino acids not able to participate in the formation of such stable complexes would have been lost to degradative processes. Perhaps not coincidentally, Root-Bernstein [
38] showed that the side chains of the same select subset of twenty amino acids can interact stereochemically across a beta ribbon, but in this case the amino acids participating in a given beta ribbon must all be of the same chirality. Once again, the interaction of the side chains across the beta ribbon would have stabilized them against degradation. The specificity of the amino acid pairings within peptides predicted by Grafstein and Root-Bernstein has subsequently been demonstrated by many research groups [
39,
40]. In addition, experiments demonstrate that a racemic mixture of serine seeded with either L- or D-polyserine will produce homochiral polymers [
41]. Thus, it seems likely that just as RNA synthesis can be chirally restrictive, so can peptide formation, and that the same kind of polymerization processes will select among available amino acids, restricting their diversity. Among these, the amino acids that could pair with specific codons (or anticodons) would again have had an additional selective benefit.
The selection for specific subsets of RNA bases and amino acids, in conjunction with the selective augmentation of concentrations of racemic mixtures of homochiral polymers of each, would have set the stage for the emergence of the genetic code and selection for homochirality within each class of compounds. Indeed, as Szathmary has suggested, it is quite possible that amino acids acted as cofactors for ribozymes, just as RNAs acted as templates for peptide synthesis, thereby creating a mutually reinforcing process of specific amino acids selecting for specific RNA sequences and vice versa [
42].
The very small number of previous experiments investigating chiral interactions between amino acids and polynucleotides has suggested, as reported here, that L-amino acids will prefer D-ribonucleotide sequences, and D-amino acids will preferentially bind to L-ribonucleotide sequences. Yarus [
11] found that a short D-ribose-based RNA sequence binds L-arginine in preference to D-arginine and Profy and Uster [
15] described non-enzymatic stereoselective aminoacylation of a D-ribose dinucleotide monophosphate selecting for L-alanine in preference to D-alanine. Tamura and Schimmel [
43,
44] have similarly demonstrated stereoselective aminoacylation of a D-polyribonucleotide sequence by L-alanine and, conversely, the stereoselectivity of the L-polyribonucleotide for D-alanine. The selectivity was about four-fold, corresponding to about 0.8 Kcals/mol binding energy, sufficient to result in a high degree of chiral purity after several rounds of selection. The experiments described above are consistent with these results. In general, L-amino acids are about two- to three-fold more likely to bind to D-oligonucleotides than are D-amino acids; and conversely, D-amino acids are about two- to three-fold more likely to bind to L-oligonucleotides than to are L-amino acids. It is therefore likely that repeated selection processes would have yielded two competing genetic codes, one comprised of L-amino acids binding to D-oligonucleotides and another of D-amino acids binding to L-oligonucleotides. There are, however, exceptions to the generalization just stated. Nolte,
et al. [
45] reported the synthesis of a 38-mer made of L-ribonucleotides that selectively bound L-Arg, and we report here that D-Cys binds preferentially to its D-codon, so the prebiotic situation may have been more complicated. Surprisingly, these exceptions may actually have contributed to the selection of an L-amino acid-D-codon code, for reasons described below.
The emergence of two, chirally selective, mirror-image codes would provide no insight into the origin of our modern code and the selection of the specific chiralities that we observe today if there were no further selection factors at work. Consider that if the four ribonucleotide bases were equally prevalent in the prebiotic environment and could polymerize with each other at identical reaction rates, then every possible short (i.e., less than about ten bases in length) polyribonucleotide sequence would have existed in the same concentrations. By the same token, if every amino acid were equally prevalent in the prebiotic environment, and the rates of polymerization of any mixture of these amino acids was identical, then every possible short peptide sequences would also exist at equal concentrations. In this case, there would be no way to select between the L-amino-acid-D-codon encodings described above, and the D-amino-acid-L-codon encodings. Neither the code nor homochirality would emerge from such as system except by the activity of an external agency such as an asymmetric force or some element of a frozen accident. We would be no further along in our search for the origins of homochirality.
If, on the other hand, there were a significant inhomogeneity in the distribution of codons (or anticodons), and a significant inhomogeneity in the distribution of amino acids, then the means for selecting between genetic codes, and thereby for homochirality, would have existed. Limited experimental evidence does exist for such inhomogeneities. Zeilenksi and Orgel [
46] reported, for example, that the oligomerization of CG is much more efficient than the oligomerization of GC. Kornberg [
47] found that oligomerization of A and T preferentially yielded
alternating AT copolymers. Thus, oligomerization does not appear to be purely random. A second skewing factor results from the observation that there are amino acid side chain interactions during peptide synthesis that result in nearest-neighbor preferences that affect peptide yields [
48]. If amino acids or short polypeptides can act as templates for codons and/or anticodons as Nelsestuen has proposed [
49,
50], then interactions between polyribonucleotides and peptides could also have resulted in significantly skewed distributions of codon and/or anticodon frequencies.
We can follow out the consequences of CG oligomerization being more efficient than that of GC with regard to the present findings [
5]. If CG oligomerizes more rapidly than GC, then under prebiotic conditions, 5’-CGU-3’ would be more prevalent than 5’-UGC-3’. According to the modern genetic code, 5’-CGU-3’ “encodes” Arg. The binding affinities reported in
Table 1 and Table 2 (and data illustrated in
Figure 5) indicate that Arg has the highest affinity for 5’-CGU-3’, as expected. Notably, the data demonstrate that L-Arg prefers D-5’-CGUA-3’ whereas D-Arg prefers L-5’-AUGC-3’ (Figure 8). Given essentially equal binding constants, equimolar concentrations of L-Arg and D-Arg, and more D-5’-CGUA-3’ than L-5’-AUGC-3’, the L-Arg- D-5’-CGUA-3’ complex would be formed in greater quantities and therefore have a kinetic advantage in any peptide polymerization reactions using it as a template. More L-Arg than D-Arg would bind to codons and be incorporated into peptides, thereby increasing the pool of L-Arg compared with D-Arg. Such a positive feedback system would promote an L-amino acid-D-codon code. Other inhomogeneities in codon production, such those described in the previous paragraph, would have yielded similar selection pressures for the modern encodings [
5].
An additional, but less obvious, selection factor would tilt the code in the same direction. This second selection factor results from the fact that multiple amino acids bind to the same codon with slight different binding constants. For example, L-methionine and D-cysteine somewhat unexpectedly both prefer D-CGUA (
Table 1 and
Figure 2), but L-Met has significantly higher affinity. Thus, in a competition between L-Met and D-Cys for D-CGUA, L-Met will “win”. One consequence of “winning” is that the concentration of free L-Met will decrease, while the concentration of D-Cys will remain high, so that D-Cys can bind to its secondarily preferred codon, which is AUGC. Once again, since GC polymerizes less rapidly than CG, the concentration of AUGC will be lower than that of CGUA, so that the L-Met-CGUA pair will be selected over the D-Cys-AUGC pair. This result is consistent with the modern genetic code, in which 5’-GUA-3’ encodes Met, not Cys.
Finally, Bolik,
et al., [
51] have provided experimental evidence that D-polyribonucleotides are more stable than L-polyribonucleotides due to small, but significant differences in their electronic structures that are evident in ultraviolet Raman spectra. An important consequence of even a slight increase in L-polyribonucleotide instability in a prebiotic system in which homochirality had not yet emerged would be to tilt the balance toward the emergence of an L-amino-acid-D-polyribonucleotide code. Notably, a problem encountered in the current experiments, which prevented testing of additional amino acid-codon pairings, was that L-AUGC began to decompose after several weeks in solution. The remaining oligomers have remained stable for several months.
Of course, the actual amount of codon-amino acid binding that occurred in such a prebiotic system would have been dependent not only on affinities but on the relative concentrations of the amino acids. Such prebiotic conditions are not mimicked here. The amino acid concentration factor probably explains why Phe, which has a higher affinity for CGUA and UAGC than do Arg, Met, Cys or Val, did not “encode” one of these codons, and that is because Phe probably has a yet higher affinity for a different codon (UUU), and was, in addition, rare compared with its competitors in the prebiotic environment in which the code evolved. Thus, it is possible to imagine conditions under which Val could “encode” 5’-CGUA-3’: such a situation would arise if the concentration of Val were several orders of magnitude higher than that of Arg. Since such a situation was not unlikely during the origins of life, it is quite possible, according to the data reported here, that every codon initially “encoded” multiple amino acids with a range of different affinities. Such multiple encodings would have enriched some amino acids through their incorporation into peptides. One can therefore imagine a scenario in which competition among amino acids for a given codon resulted in selection not of a single amino acid, but of any one of a small set (say, two to four) amino acids. While such a “sloppy” translation system would result in a wide range of peptide products, the result of many iterations of such a process would be a net increase in the amino acids associated with that codon and elimination of those that were not. Given the kind of overlapping encodings observed here, the result would have been a significant pruning of RNA base diversity as well as amino acid diversity.
In sum, the data presented here demonstrate that it is quite possible that homochirality was not established at the time the genetic code evolved, but that a general preference of L-amino acids for D-codons and D-amino acids for L-codons, combined with inhomogeneities in the distribution and concentrations of amino acids and codons, led to the selection of the current genetic code. The operative factor in the selection of the modern genetic code was competition among L- and D-amino acids for L- and D-oligonucleotides. L-amino acids tend to prefer D-codons while D-amino acids prefer L-codons. Given what is currently known about the kinetics of oligimerization and probable prebiotic amino acid prevalence, the data presented here would lead to selection of an L-amino acid-D-codon code with the general set of encodings found in the modern genetic code.
It is important to stress that the outcome of such a selection process was not a foregone conclusion [
5]. Under conditions in which some amino acids that were relatively rare on earth were more common, and/or the relative concentrations of adenine and uracil were significantly greater than cytosine and guanine (or different bases were selected altogether), then an entirely different code might have emerged. Under such circumstances, it might also have been possible for a D-amino acid-L-codon code to be selected. We should not, therefore, assume that life on other planets or in other solar systems necessarily shares the genetic encodings that we have on earth, nor that extraterrestrial life will share our homochiralities.
Finally, we note that some means of amplifying the small selection pressures described here would have been necessary for the emergence of an unambiguous genetic code comprised of strict homochiral components. It is extremely important that such enrichment studies be carried out over many iterations to determine how competition among the various possible encodings would play out in mixtures of amino acids at concentrations modeling possible prebiotic scenarios. Are the two- to three-fold differences in affinities combined with inhomogeneities in codon availability sufficient to yield L-amino-acid-D-codon domination as suggested here? Or may other forms of chiral amplification required? Many possibilities for such enrichment certainly exist of which simple repetitive selection cycles are only one. As Tamura and Schimmel [
43,
44] have pointed out with regard to their own research, a four-fold difference in binding affinity can result in nearly complete selection for homochirality within a half-dozen selection cycles. Since the difference in affinity are somewhat less in the data reported here, it might take a dozen or more cycles to achieve relative purity, but this prediction is testable. Secondly, it is notable that previous research on the amino acylation of D-RNAs in solution has not yielded consistent evidence of a chiral preference for amino acids, but when the RNAs are immobilized on a surface (as might have occurred during the evolution of life), a clear preference for L-amino acids was manifested [
52]. It is possible that immobilization of the oligonucleotides tested here might significantly increase their chiral preferences as well, which might strengthen the case for the emergence of homochirality with the genetic code considerably. And lastly, there are a very wide range of chiral amplification processes that have been characterized besides those just mentioned, that include isotactic polymer formation [
27,
30,
53,
54]; lattice formation on surfaces such as clays [
55]; non-linear thermodynamic asymmetric amplification of amino acid synthesis [
56]; peptide-peptide binding, in which peptides composed of L-amino acids would preferentially bind to other L-based peptides, while D-amino acid-based peptides would prefer those composed of D-amino acids [
38,
39,
57,
58]; peptide replication [
59,
60,
61] in which an existing L-rich peptide would preferentially act as a template for further L-rich complementary peptides; and multiple levels of selection including formation of RNA-amino acid conjugates [
62].
In sum, the current investigation of L- and D-amino acid binding to L- and D- oligonucleotide sequences has helped to confirm the theory that the genetic code emerged through direct amino acid-codon interactions [
5]. Notably, however, every codon seems to have a range of affinities for a variety of amino acids, so that competition for genetic encodings must be assumed to have occurred. New and much more extensive data has been provided demonstrating that L-amino acids generally (but not always!) prefer D-oligonucleotides, while D-amino acids generally prefer L-oligonucleotides. There is some amino acid preference for codon directionality, but again, codon directionality may not have been a foregone conclusion during the emergence of the genetic code. Finally, the data strongly suggest that at least two competing genetic codes would have emerged during evolution, and that experimentally demonstrated forms of inhomogeneities in codon production and stability were the likely factors that tipped the competition between these codes toward the L-amino-acid-D-codon system that we have today. Thus, the testing of the co-evolution of homochirality and code (COHAC) hypothesis has yielded novel, perhaps surprising, and hopefully useful results. Further research involving multiple cycles of amino acid-codon selection, binding studies involving surface-bound oligonucleotides, as well as other forms of chiral amplification, may clarify the actual mechanisms by which homochirality may have emerged with the genetic code.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}