Urea Memory: Transient Cell Exposure to Urea Causes Persistent Mitochondrial ROS Production and Endothelial Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Transient Exposure of Endothelial Cells to a Disease-Relevant Concentration of Urea Induces Increased Mitochondrial ROS Production That Persists for Days after Urea Is Removed
2.2. Cytosolic and Mitochondrial ROS Generating Mechanisms Initiate the Persistent ROS Production Induced by Transient Exposure to 20 mM Urea in Endothelial Cells
2.3. Urea-Induced Mitochondrial Dysfunction Maintains Persistent Mitochondrial ROS Production in Endothelial Cells after Transient Exposure to High Urea Concentrations
2.3.1. Transient Exposure to 20 mM Urea Induces a Long-Lasting Reduction of mtDNA Copy Number in Endothelial Cells
2.3.2. Transient Exposure to 20 mM Urea Causes a Long-Lasting Reduction of Electron Transport Chain Component Expression
2.3.3. Transient Exposure to 20 mM Urea Causes a Long-Lasting Dysregulation of Mitochondrial Dynamics in Human Aortic Endothelial Cells
2.3.4. Transient HU Causes Endothelial Cell Dysfunction Lasting for 2 Days Despite Urea Normalization
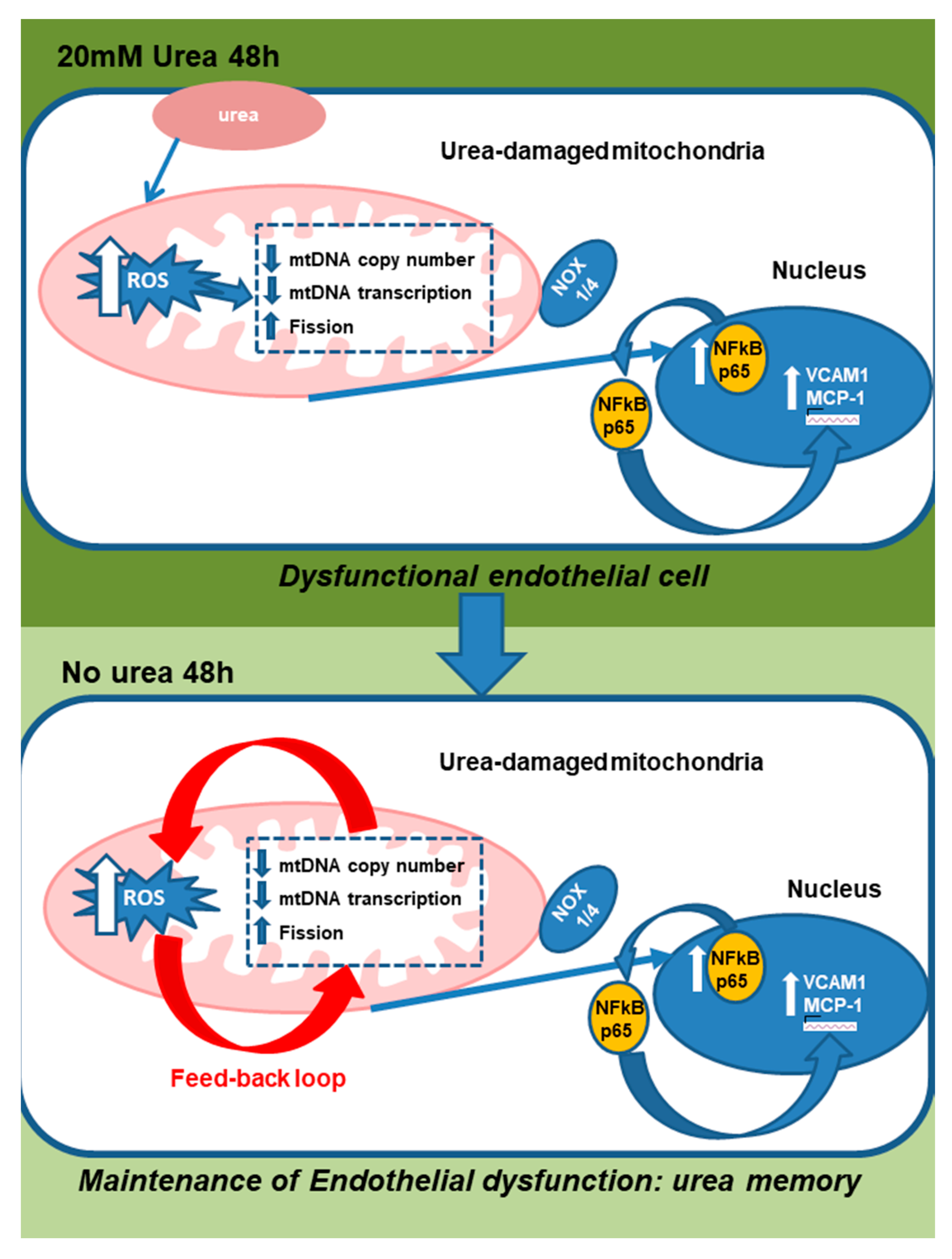
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions
4.2. Adenoviral Vectors
4.3. NADPH Oxidase Inhibition
4.4. Measurement of ROS Generation
4.5. Determination of mtDNA Content by Real-Time Quantitative Polymerase Chain Reaction
4.6. RT Reaction and Real-Time Quantitative PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Nonstandard Abbreviations
| CRF | chronic renal failure |
| ROS | reactive oxygen species |
| ESRD | end stage renal disease |
| CVD | cardiovascular disease |
| HU | High urea; NU No urea |
References
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Tonelli, M.; Karumanchi, S.A.; Thadhani, R. Epidemiology and Mechanisms of Uremia-Related Cardiovascular Disease. Circulation 2016, 133, 518–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, H.; Nishizawa, Y. AGEs/RAGE in CKD: Irreversible metabolic memory road toward CVD? Eur. J. Clin. Investig. 2010, 40, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Drüeke, T.B.; Massy, Z.A. Atherosclerosis in CKD: Differences from the general population. Nat. Rev. Nephrol. 2010, 6, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Harmankaya, O.; Akalin, N.; Akay, H.; Okuturlar, Y.; Erturk, K.; Kaptanogullari, H.; Kocoglu, H. Comparison of risk factors for cardiovascular disease in hemodialysis and peritoneal dialysis patients. Clinics 2015, 70, 601–605. [Google Scholar] [CrossRef]
- Golestaneh, L.; Melamed, M.L.; Hostetter, T.H. Uremic memory: The role of acute kidney injury in long-term outcomes. Kidney Int. 2009, 76, 813–814. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Yunlei, D.; Qiuling, F.; Xu, W.; Qianwen, Z.; Xu, C.; Li, X.; Lining, W. Transient High-Glucose Stimulation Induces Persistent Inflammatory Factor Secretion from Rat Glomerular Mesangial Cells via an Epigenetic Mechanism. Cell Physiol. Biochem. 2018, 49, 1747–1754. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Du, X.; Carratú, A.; Gerfen, G.J.; D’Apolito, M.; Giardino, I.; Rasola, A.; Marin, O.; Divakaruni, A.S.; Murphy, A.N.; et al. GLP-1 Cleavage Product Reverses Persistent ROS Generation after Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes 2015, 64, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Apolito, M.; Colia, A.L.; Lasalvia, M.; Capozzi, V.; Falcone, M.P.; Pettoello-Mantovani, M.; Brownlee, M.; Maffione, A.B.; Giardino, I. Urea-induced ROS accelerate senescence in endothelial progenitor cells. Atherosclerosis 2017, 263, 127–136. [Google Scholar] [CrossRef] [PubMed]
- D’Apolito, M.; Du, X.; Zong, H.; Catucci, A.; Maiuri, L.; Trivisano, T.; Pettoello-Mantovani, M.; Campanozzi, A.; Raia, V.; Pessin, J.E.; et al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J. Clin. Investig. 2010, 120, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukaiand, M. NAD(P)H Oxidase Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of “Free Radical Diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar] [PubMed]
- Giedt, R.J.; Yang, C.; Zweier, J.L.; Matzavinos, A.; Alevriadou, B.R. Mitochondrial fission in endothelial cells following simulated ischemia/reperfusion: Role of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 2012, 52, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chan, D.C. OPA1 and cardiolipin team up for mitochondrial fusion. Nat. Cell Biol. 2017, 19, 760–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Lüscher, B.; Hottiger, M.O. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 15, 4251–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantino, S.; Paneni, F.; Cosentino, F. Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr. Pharm. Biotechnol. 2015, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia Induces a Dynamic Cooperativity of Histone Methylase and Demethylase Enzymes Associated with Gene-Activating Epigenetic Marks That Coexist on the Lysine Tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Neddermann, D.; Piorunska-Stolzmann, M.; Jagodzinski, P.P. Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res. Clin. Pract. 2014, 105, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Pietrement, C.; Touré, F. Reconsidering the Lack of Urea Toxicity in Dialysis Patients. Semin. Dial. 2016, 29, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Giardino, I.; D’Apolito, M.; Brownlee, M.; Maffione, A.B.; Colia, A.L.; Sacco, M.; Ferrara, P.; Pettoello-Mantovani, M. Vascular toxicity of urea, a new “old player” in the pathogenesis of chronic renal failure induced cardiovascular diseases. Turk. Arch. Pediatr. 2017, 52, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.M.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; Shanske, S.; Tritschler, H.J.; Aprille, J.R.; Andreetta, F.; Bonilla, E.; Schon, E.A.; DiMauro, S. mtDNA depletion with variable tissue expression a novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet. 1991, 48, 492–501. [Google Scholar] [PubMed]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in Atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Mercer, J.; Bennett, M. Mitochondria in vascular disease. Cardiovasc. Res. 2012, 95, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamboa, J.L.; Billings, F.T., IV; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J., II; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, C.Y.; Park, J.T.; Kee, Y.K.; Han, S.G.; Han, I.M.; Kwon, Y.E.; Park, K.S.; Lee, M.J.; Han, S.H.; Kang, S.W.; et al. Low Mitochondrial DNA Copy Number is Associated with Adverse Clinical Outcomes in Peritoneal Dialysis Patients. Medicine 2016, 95, e2717. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Massy, Z.; Argiles, A.; Spasovski, G.; Verbeke, F.; Lameire, N.; European Uremic Toxin Work Group. Chronic kidney disease as a cause of cardiovascular morbidity and mortality. Nephrol. Dial. Transpl. 2005, 20, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Al-Kafaji, G.; Golbahar, J. High glucose-induced oxidative stress increases the copy number of mitochondrial DNA in human mesangial cells. Biomed. Res. Int. 2013, 2013, 754946. [Google Scholar] [CrossRef] [PubMed]


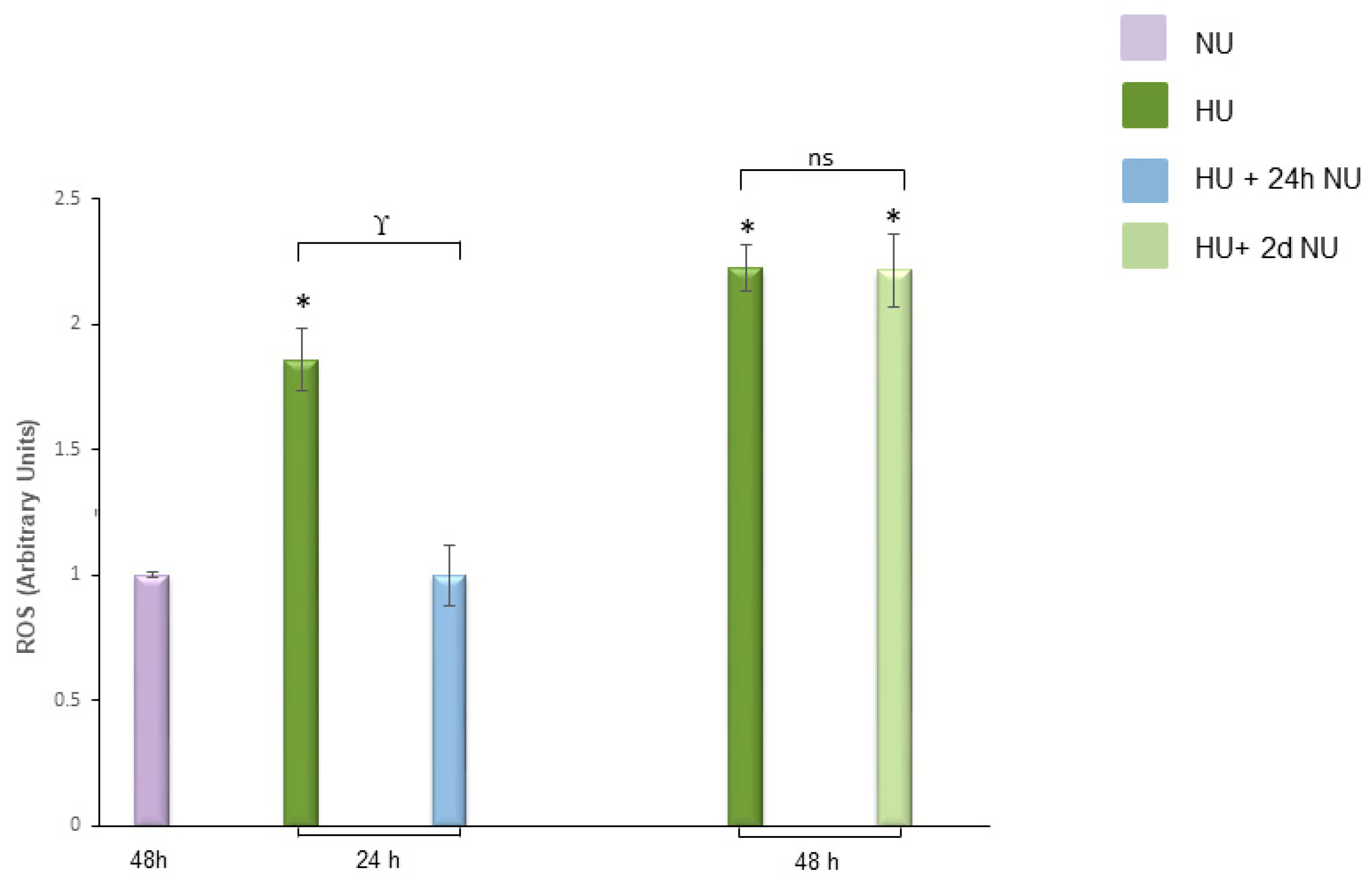
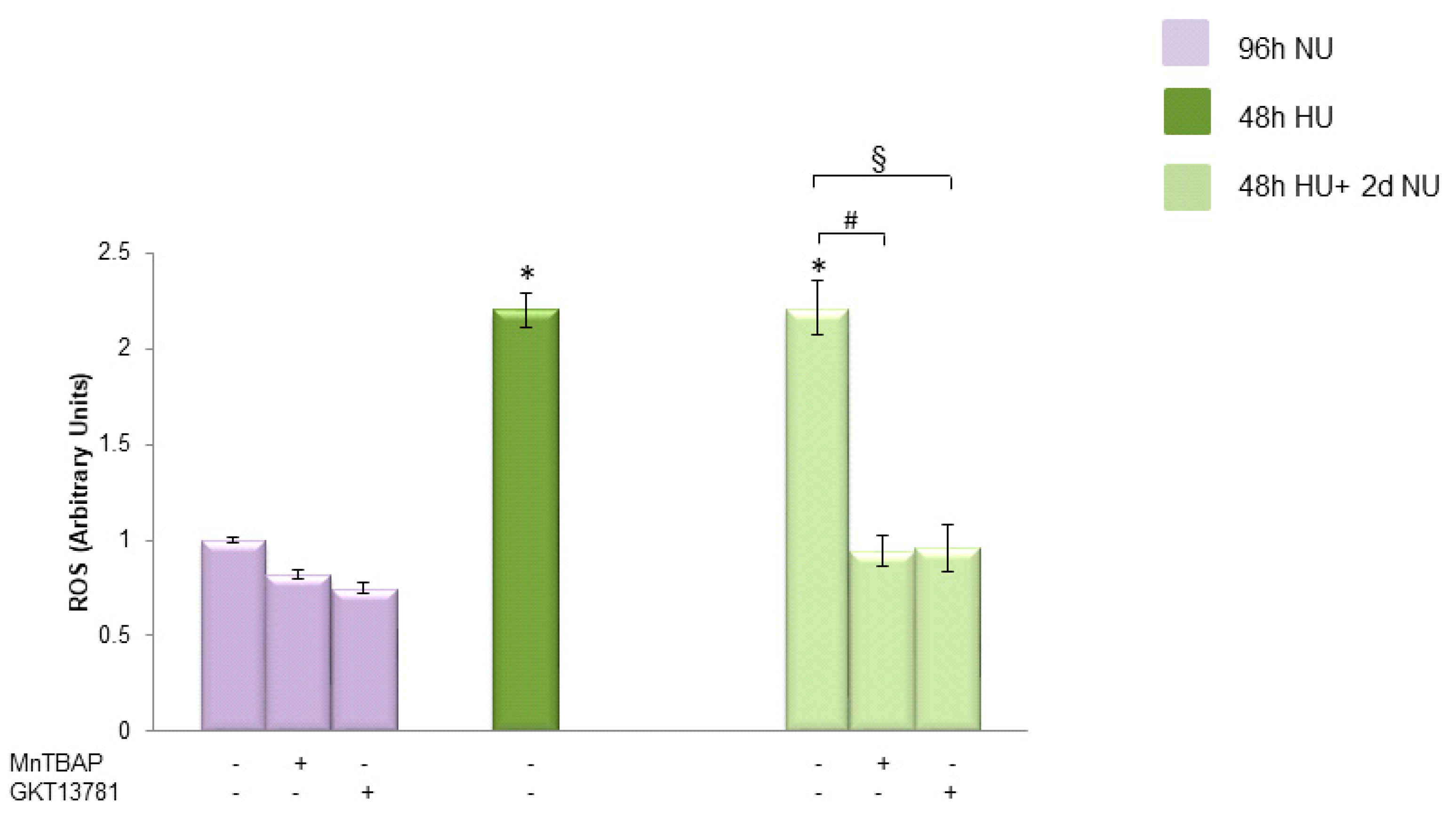
 , and to 20 mM urea for 48 h followed by two days of subsequent incubation in media without urea (NU)
, and to 20 mM urea for 48 h followed by two days of subsequent incubation in media without urea (NU)  ± MnTBAP or GKT13781. ROS levels were measured by CM-H2DCFDA. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitors compared to cells treated with MnTBAP, § p < 0.05 cells not treated with the inhibitors compared to cells treated with GKT13781.
, and to 20 mM urea for 48 h followed by two days of subsequent incubation in media without urea (NU) ± MnTBAP or GKT13781. ROS levels were measured by CM-H2DCFDA. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitors compared to cells treated with MnTBAP, § p < 0.05 cells not treated with the inhibitors compared to cells treated with GKT13781.
± MnTBAP or GKT13781. ROS levels were measured by CM-H2DCFDA. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitors compared to cells treated with MnTBAP, § p < 0.05 cells not treated with the inhibitors compared to cells treated with GKT13781.
, and to 20 mM urea for 48 h followed by two days of subsequent incubation in media without urea (NU) ± MnTBAP or GKT13781. ROS levels were measured by CM-H2DCFDA. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitors compared to cells treated with MnTBAP, § p < 0.05 cells not treated with the inhibitors compared to cells treated with GKT13781.
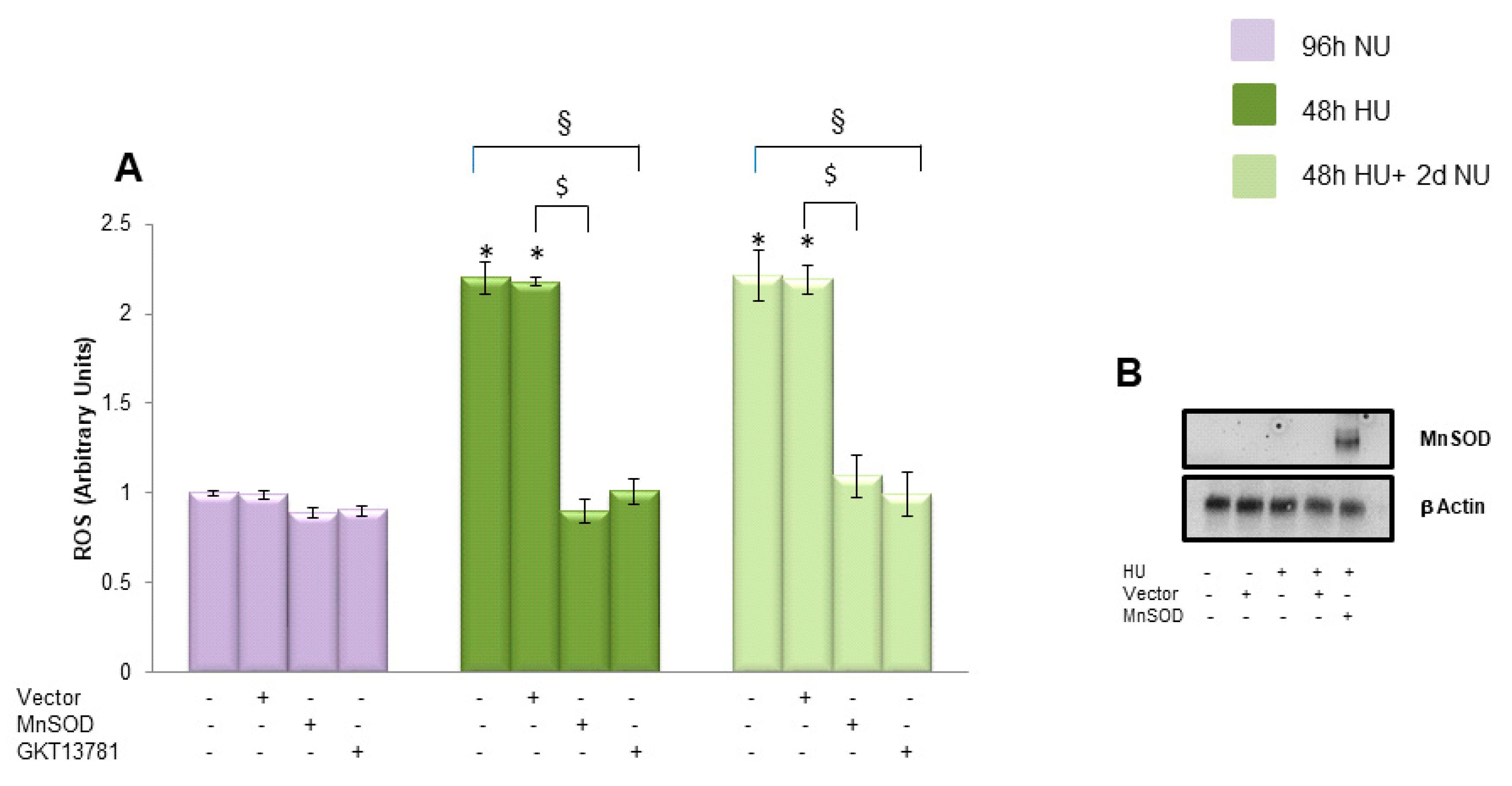
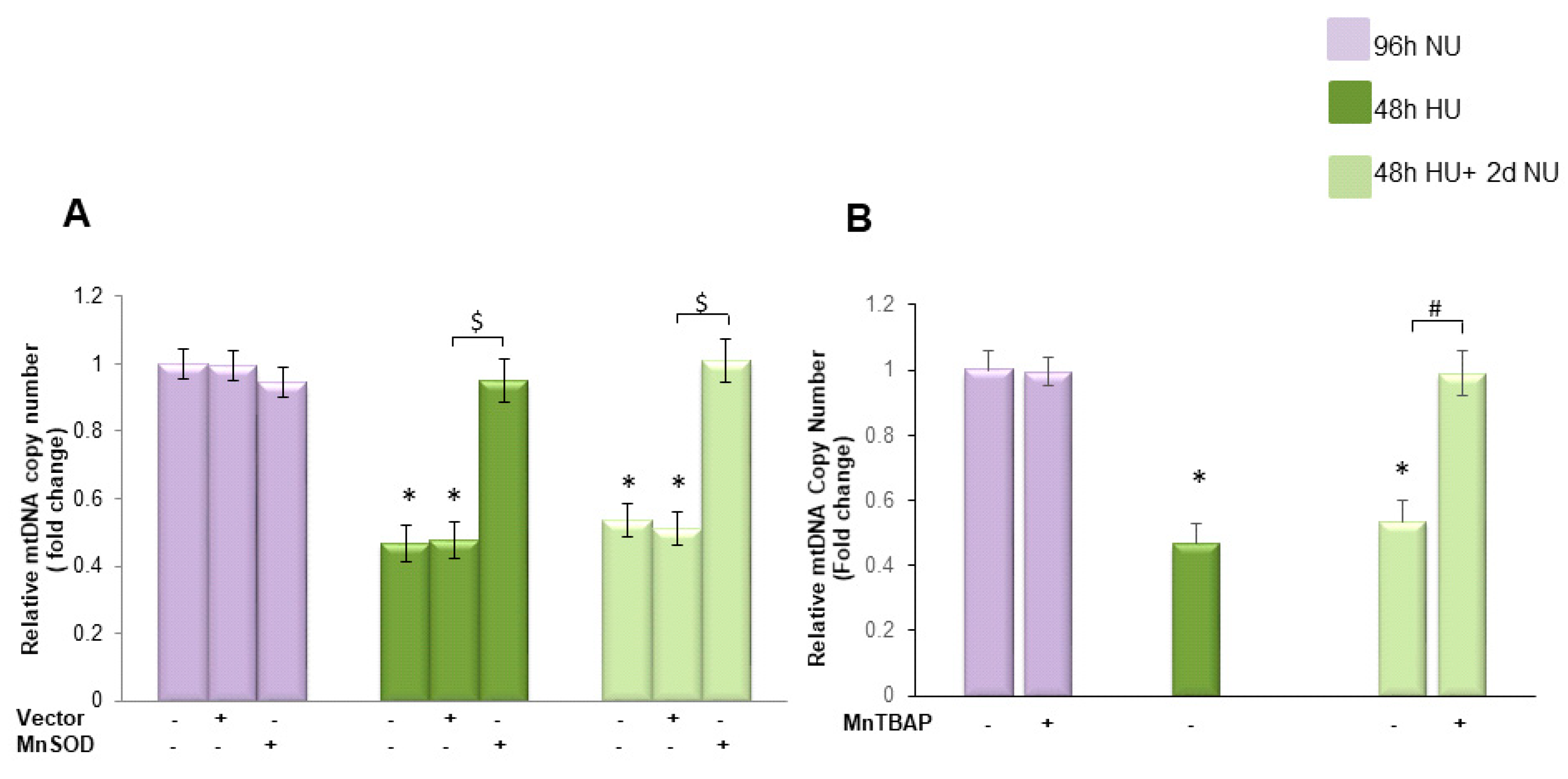
 , and to 20 mM urea followed by two days of subsequent incubation in media without urea (NU)
, and to 20 mM urea followed by two days of subsequent incubation in media without urea (NU)  . In the indicated groups, cells were infected with MnSOD before transient exposure to urea (A), or were treated with MnTBAP during the two days of subsequent incubation in media alone (B). mtDNA copy number was measured by a quantitative Real-time polymerase chain reaction (PCR)-based method. Data are the mean ± S.E. from seven independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
, and to 20 mM urea followed by two days of subsequent incubation in media without urea (NU) . In the indicated groups, cells were infected with MnSOD before transient exposure to urea (A), or were treated with MnTBAP during the two days of subsequent incubation in media alone (B). mtDNA copy number was measured by a quantitative Real-time polymerase chain reaction (PCR)-based method. Data are the mean ± S.E. from seven independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
. In the indicated groups, cells were infected with MnSOD before transient exposure to urea (A), or were treated with MnTBAP during the two days of subsequent incubation in media alone (B). mtDNA copy number was measured by a quantitative Real-time polymerase chain reaction (PCR)-based method. Data are the mean ± S.E. from seven independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
, and to 20 mM urea followed by two days of subsequent incubation in media without urea (NU) . In the indicated groups, cells were infected with MnSOD before transient exposure to urea (A), or were treated with MnTBAP during the two days of subsequent incubation in media alone (B). mtDNA copy number was measured by a quantitative Real-time polymerase chain reaction (PCR)-based method. Data are the mean ± S.E. from seven independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
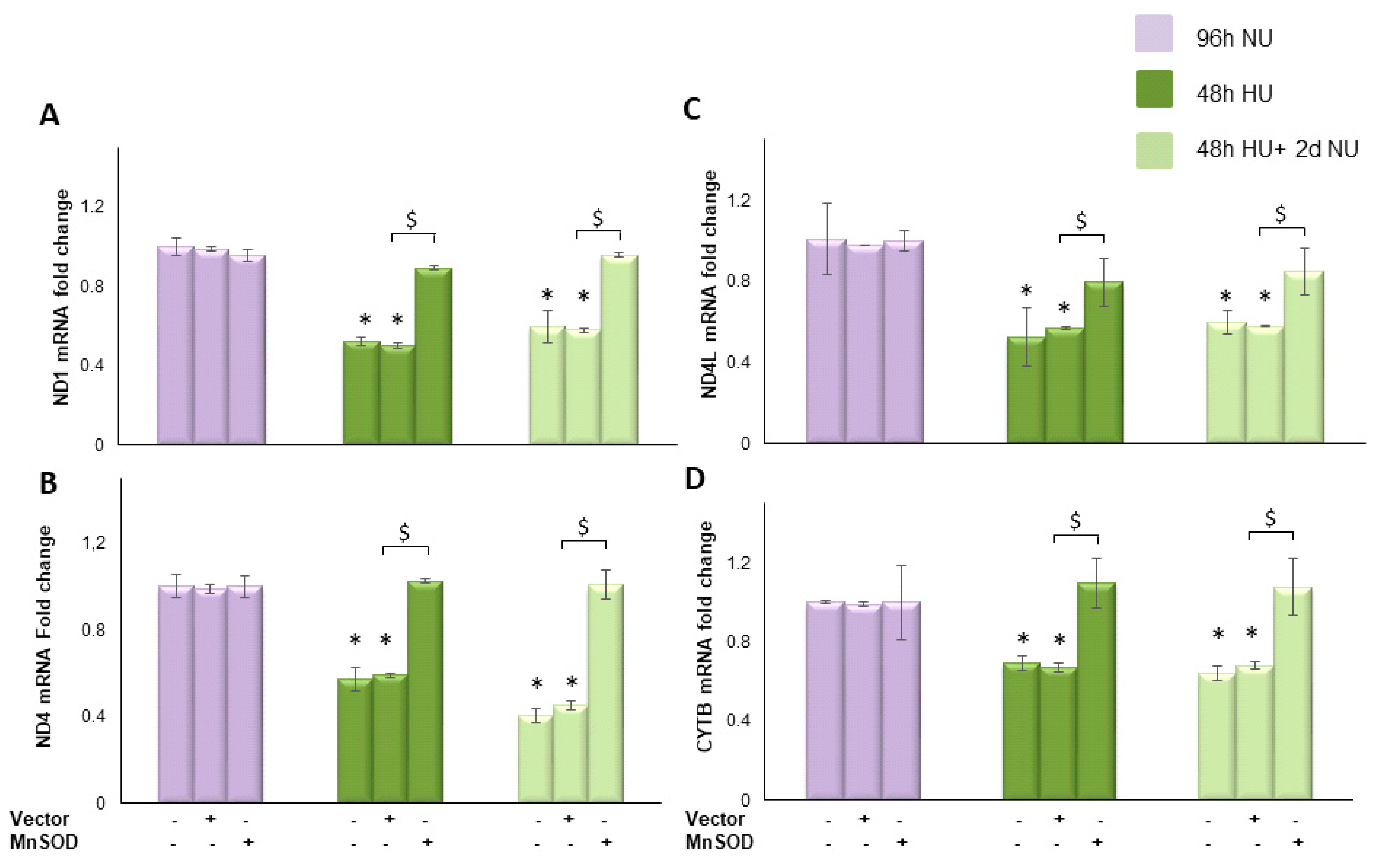
 , or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone
, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone  . The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD.
, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone . The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD.
. The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD.
, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone . The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, $ p < 0.05 cells transfected with the empty vector compared to cells over expressing MnSOD.
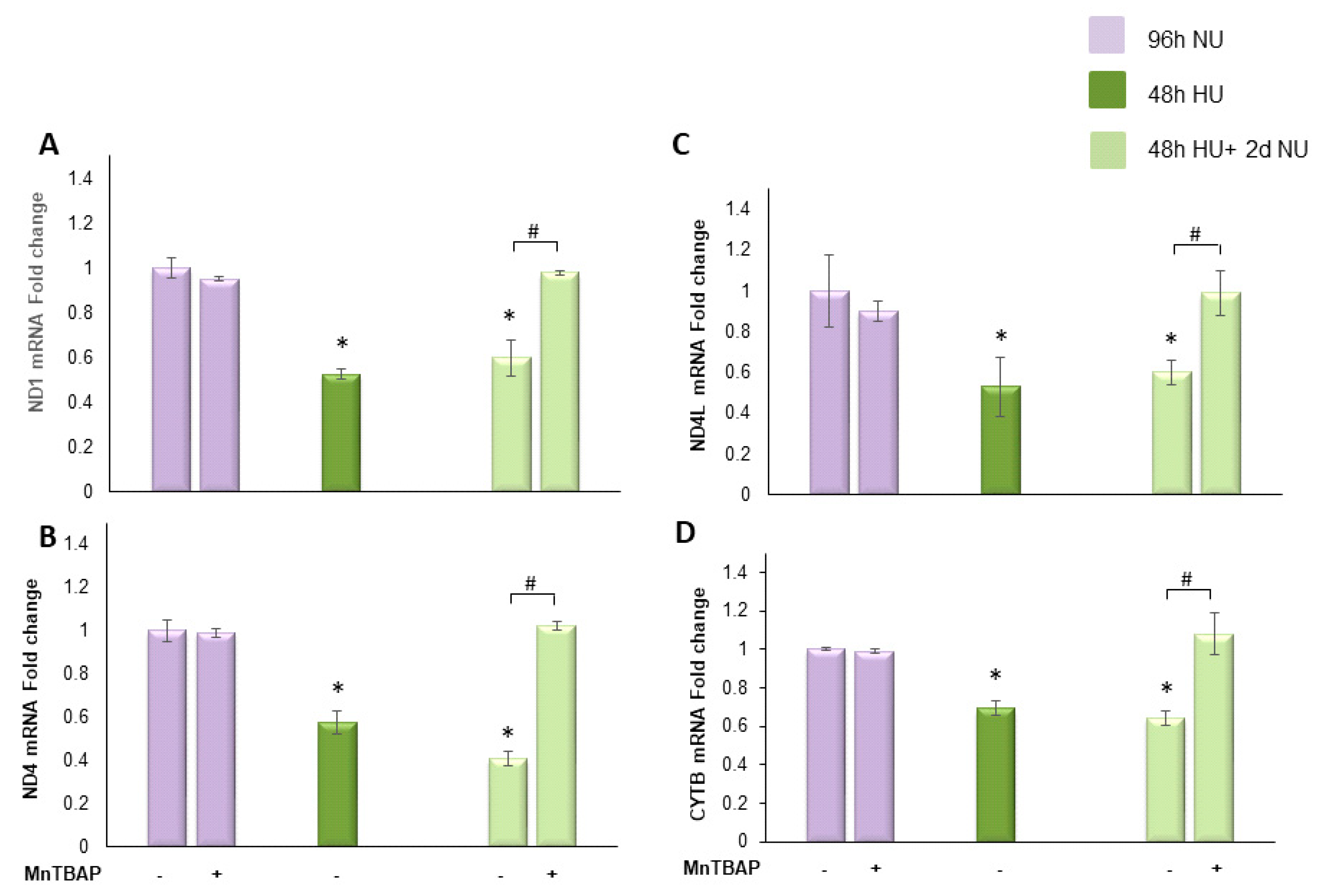
 for 48 h, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone,
for 48 h, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone,  , with or without MnTBAP. The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
for 48 h, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone, , with or without MnTBAP. The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
, with or without MnTBAP. The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
for 48 h, or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone, , with or without MnTBAP. The mRNA expression was measured by Real time PCR: (A) ND1; (B) ND4; (C) ND4L; and (D) cytochrome b. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.

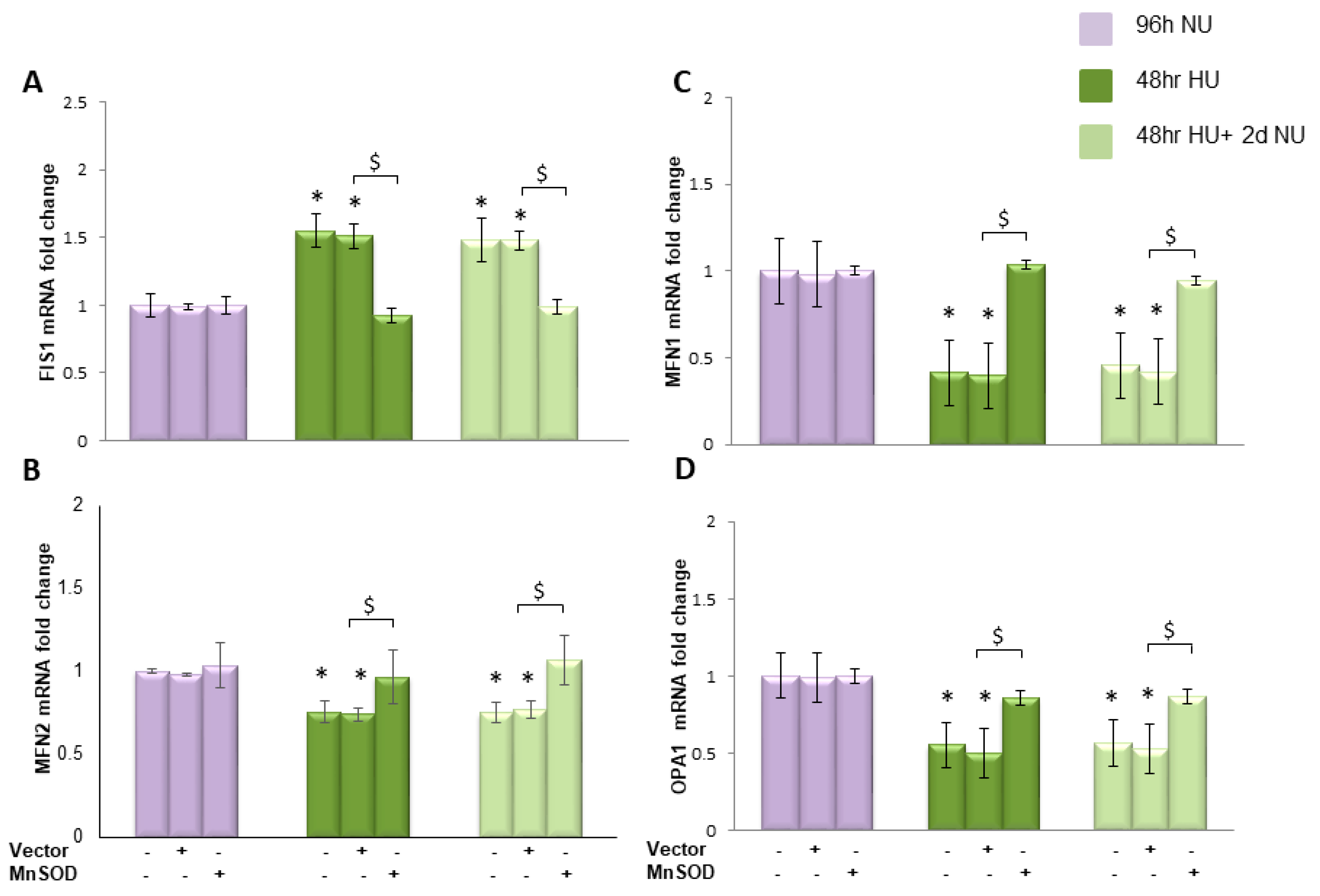
 or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone, with or without MnTBAP
or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone, with or without MnTBAP  . The mRNA expression was measured by Real time PCR: (A) Fis1; (B) Mfn1; (C) Mfn2; and (D) OPA1. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone, with or without MnTBAP . The mRNA expression was measured by Real time PCR: (A) Fis1; (B) Mfn1; (C) Mfn2; and (D) OPA1. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
. The mRNA expression was measured by Real time PCR: (A) Fis1; (B) Mfn1; (C) Mfn2; and (D) OPA1. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone, with or without MnTBAP . The mRNA expression was measured by Real time PCR: (A) Fis1; (B) Mfn1; (C) Mfn2; and (D) OPA1. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
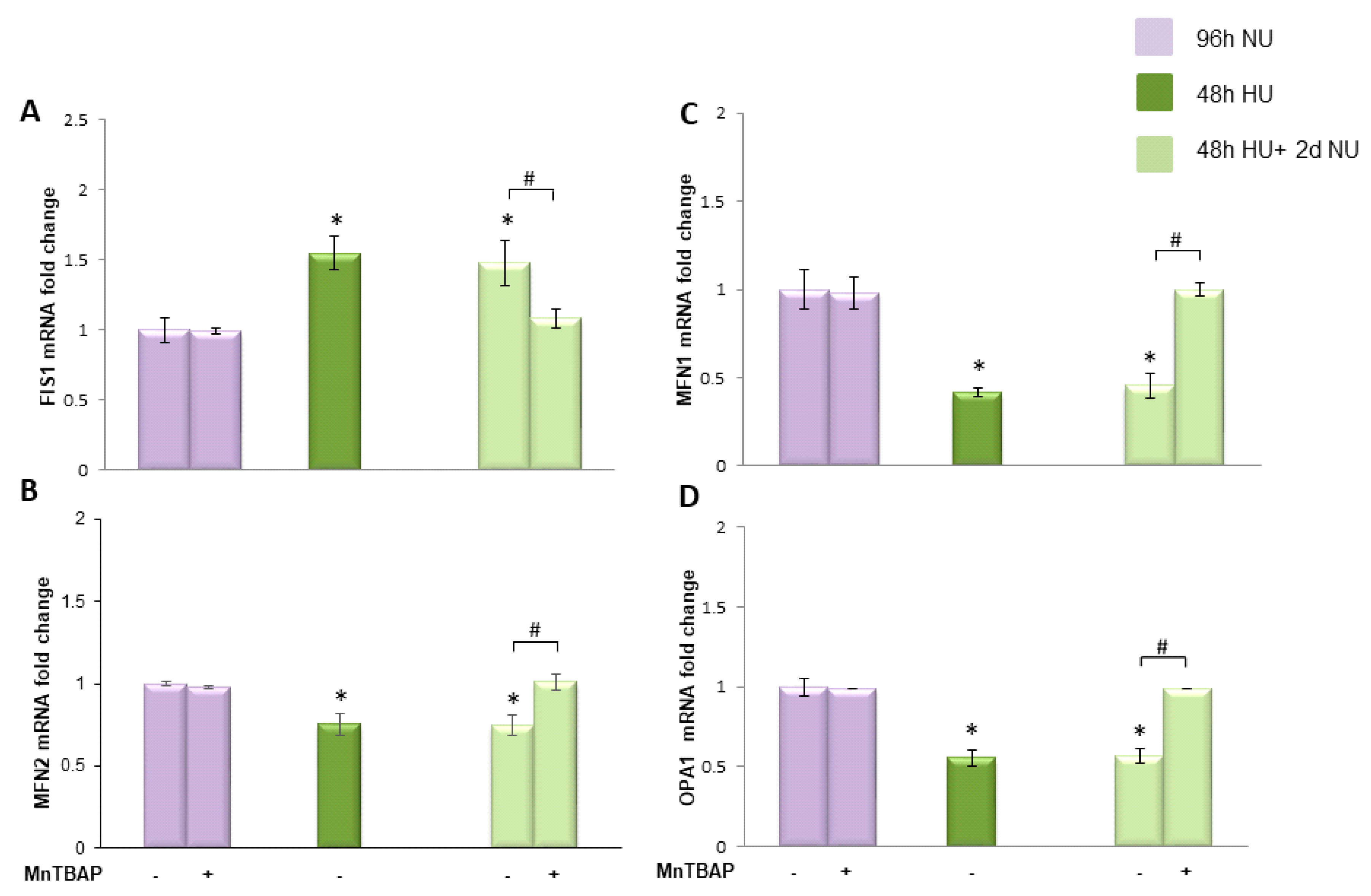
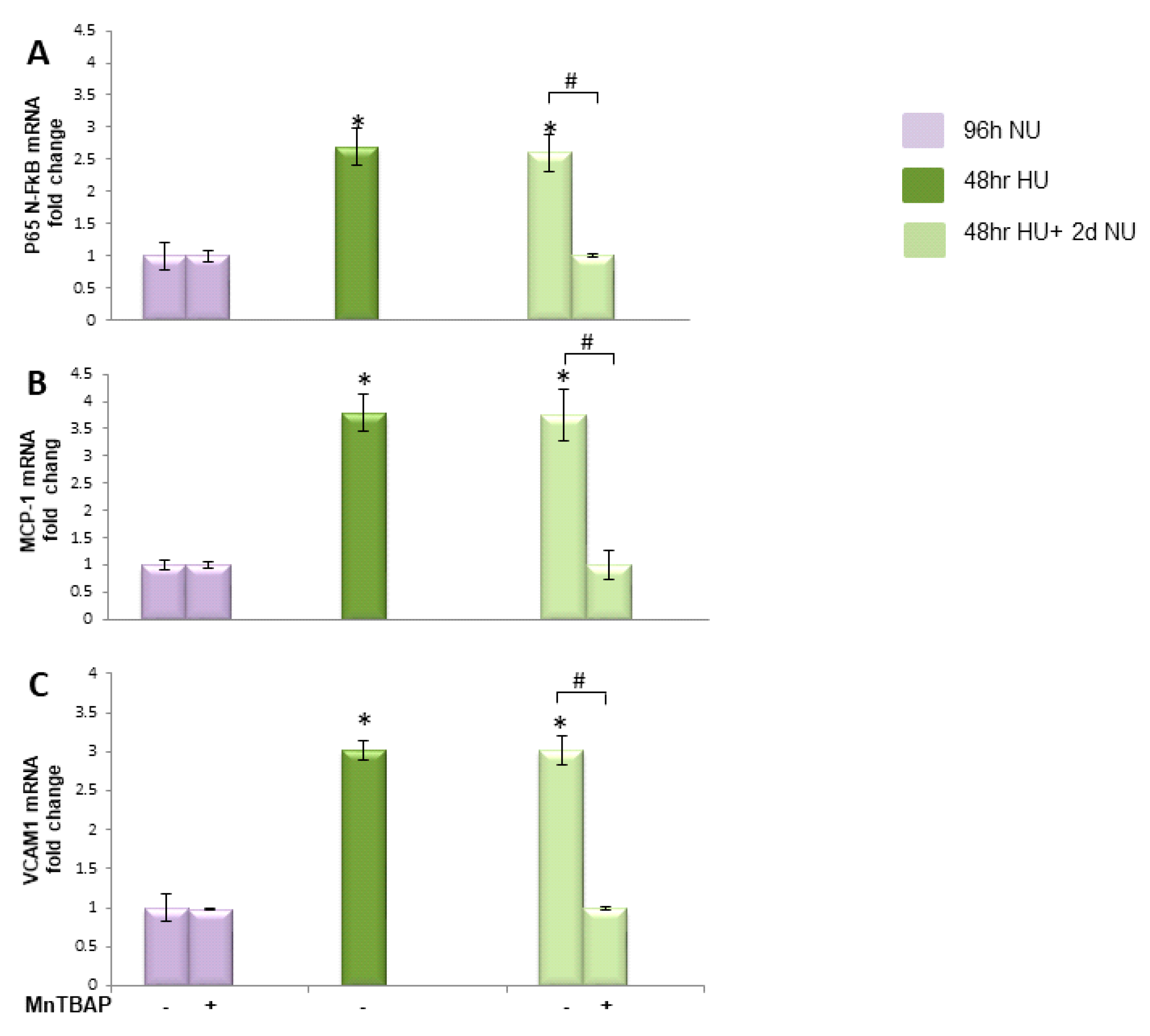
 or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone
or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone  ± MnTBAP. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone ± MnTBAP. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
± MnTBAP. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.
or to 20 mM urea for 48 h followed by two days of subsequent incubation in media alone ± MnTBAP. Data are the mean ± S.E. from five independent experiments. * p < 0.05 compared to cells treated in the same conditions but not exposed to urea, # p < 0.05 cells not treated with the inhibitor compared to cells treated with MnTBAP.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Apolito, M.; Colia, A.L.; Manca, E.; Pettoello-Mantovani, M.; Sacco, M.; Maffione, A.B.; Brownlee, M.; Giardino, I. Urea Memory: Transient Cell Exposure to Urea Causes Persistent Mitochondrial ROS Production and Endothelial Dysfunction. Toxins 2018, 10, 410. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100410
D’Apolito M, Colia AL, Manca E, Pettoello-Mantovani M, Sacco M, Maffione AB, Brownlee M, Giardino I. Urea Memory: Transient Cell Exposure to Urea Causes Persistent Mitochondrial ROS Production and Endothelial Dysfunction. Toxins. 2018; 10(10):410. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100410
Chicago/Turabian StyleD’Apolito, Maria, Anna Laura Colia, Enrica Manca, Massimo Pettoello-Mantovani, Michele Sacco, Angela Bruna Maffione, Michael Brownlee, and Ida Giardino. 2018. "Urea Memory: Transient Cell Exposure to Urea Causes Persistent Mitochondrial ROS Production and Endothelial Dysfunction" Toxins 10, no. 10: 410. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins10100410