Multiple CH/π Interactions Maintain the Binding of Aflatoxin B1 in the Active Cavity of Human Cytochrome P450 1A2


Abstract
:1. Introduction
2. Results and Discussion
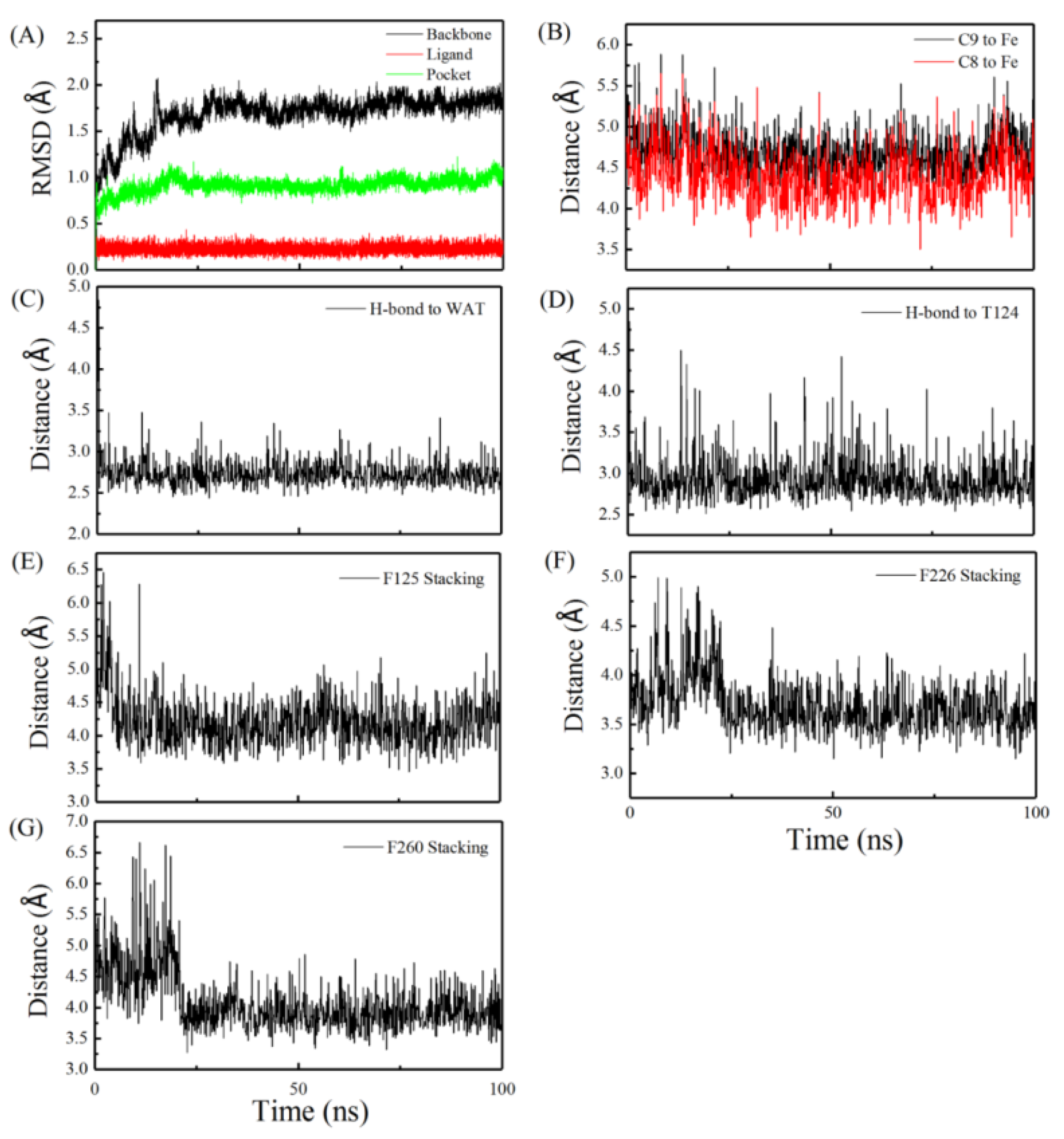
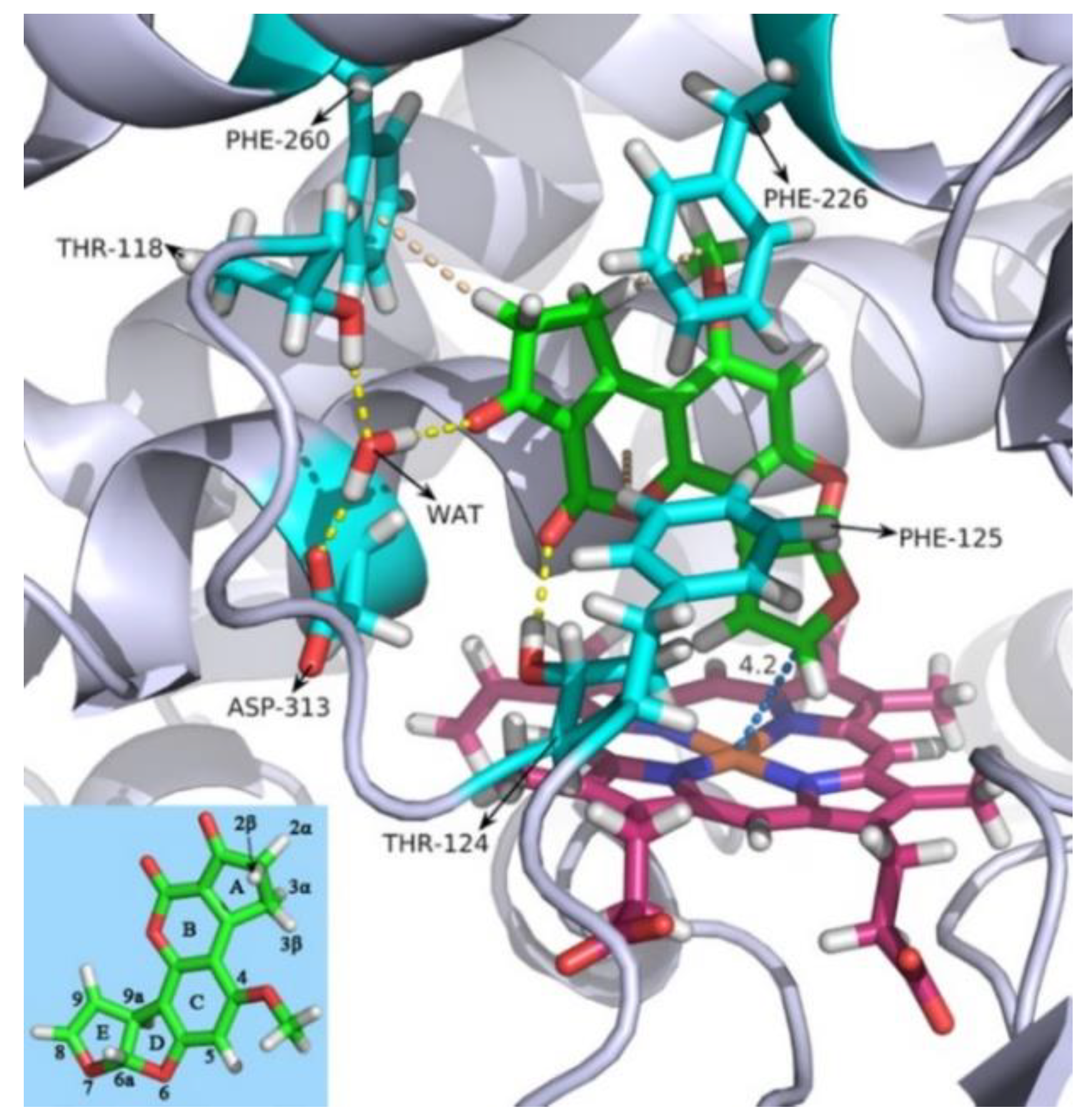
2.1. Molecular Dynamics Simulation
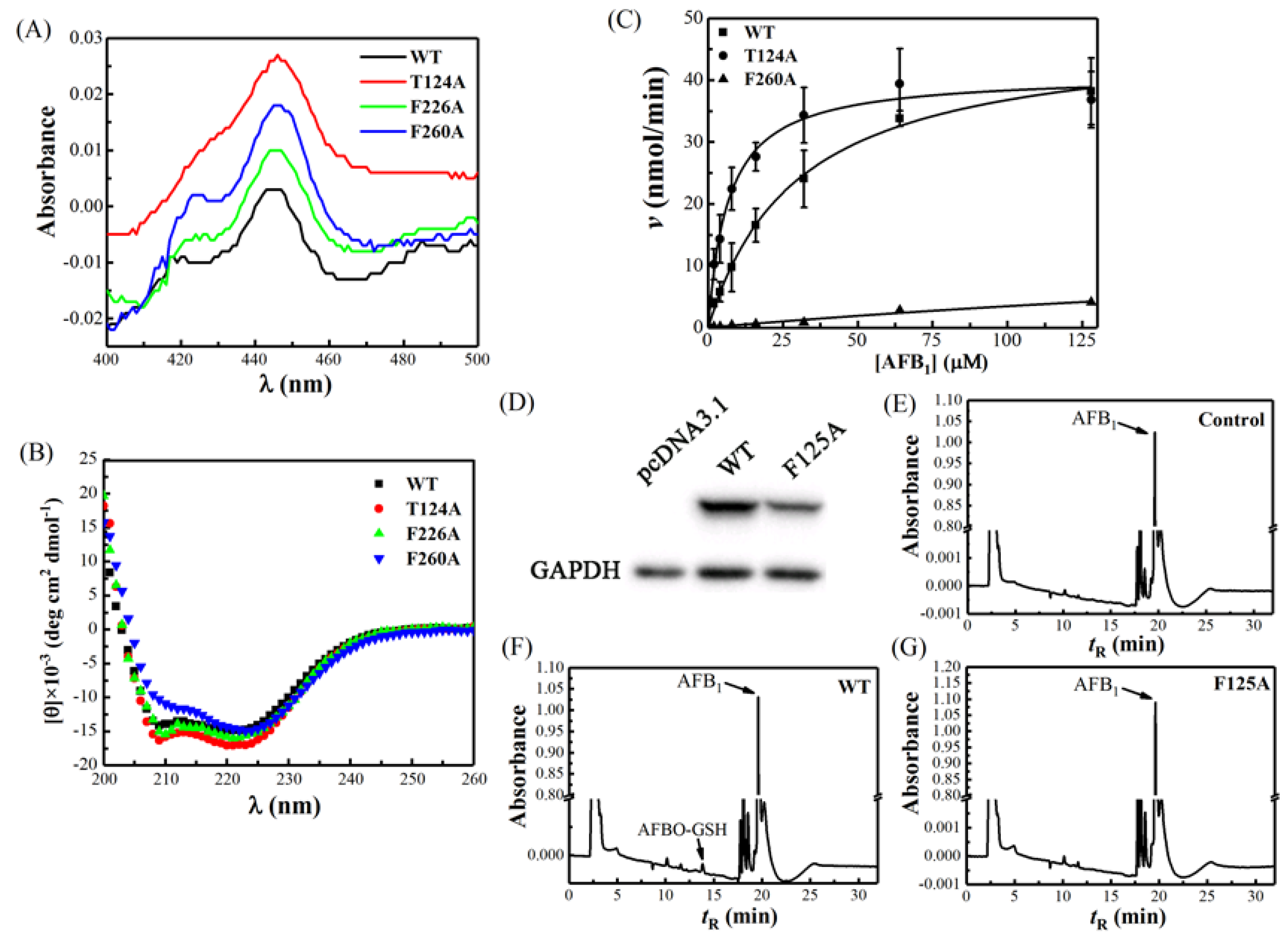
2.2. The Spectral Property of Escherichia coli-Expressed CYP Proteins
2.3. Activity Analysis
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Molecular Docking
4.3. MD Simulation
4.4. Vector Construction and Site-Directed Mutagenesis
4.5. Protein Expression and Purification
4.6. Cell Assays
4.7. Immunoblotting Analysis
4.8. CO Difference Spectra
4.9. Circular Dichroism Spectroscopy
4.10. Activity Assays
4.11. HPLC Assays
4.12. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lonsdale, R.; Fort, R.M.; Rydberg, P.; Harvey, J.N.; Mulholland, A.J. Quantum mechanics/molecular mechanics modeling of drug metabolism: Mexiletine N-hydroxylation by cytochrome P450 1A2. Chem. Res. Toxicol. 2016, 29, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenquist, T.A.; Einolf, H.J.; Dickman, K.G.; Wang, L.; Smith, A.; Grollman, A.P. Cytochrome P450 1A2 detoxicates aristolochic acid in the mouse. Drug Metab. Dispos. 2010, 38, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Szklarz, G.D. Significant increase in phenacetin oxidation on L382V substitution in human cytochrome P450 1A2. Drug Metab. Dispos. 2010, 38, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Bergstrom, R.F.; Reddy, S.; Quinlan, T.; Chappell, J.; Hong, Q.; Ring, B.; Knadler, M.P. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharmacokinet. 2008, 47, 191–202. [Google Scholar] [CrossRef]
- Kot, M.; Daniel, W.A. The relative contribution of human cytochrome P450 isoforms to the four caffeine oxidation pathways: An in vitro comparative study with cDNA-expressed P450s including CYP 2C isoforms. Biochem. Pharmacol. 2008, 76, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17 β-estradiol and estrone formed by 15 selectively expressed human cytochrome P450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef] [PubMed]
- Zaccaro, C.; Sweitzer, S.; Pipino, S.; Gorman, N.; Sinclair, P.R.; Sinclair, J.F.; Nebert, D.W.; De Matteis, F. Role of cytochrome P450 1A2 in bilirubin degradation studies in CYP1A2 (-/-) mutant mice. Biochem. Pharmacol 2001, 61, 843–849. [Google Scholar] [CrossRef]
- Kim, D.; Guengerich, F.P. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.P.; Wienkers, L.C.; Stapleton, P.L.; Kunze, K.L.; Eaton, D.L. Role of human microsomal and human complementary DNA-expressed cytochromes P450 1A2 and P450 3A4 in the bioactivation of aflatoxin B1. Cancer Res. 1994, 54, 101–108. [Google Scholar]
- Kensler, T.W.; Roebuck, B.D.; Wogan, G.N.; Groopman, J.D. Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci. 2011, 120, S28–S48. [Google Scholar] [CrossRef]
- Groopman, J.D.; Kensler, T.W. Role of metabolism and viruses in aflatoxin-induced liver cancer. Toxicol. Appl. Pharmacol. 2005, 206, 131–137. [Google Scholar] [CrossRef]
- Kamdem, L.K.; Meineke, I.; Godtel-Armbrust, U.; Brockmoller, J.; Wojnowski, L. Dominant contribution of P450 3A4 to the hepatic carcinogenic activation of aflatoxin B1. Chem. Res. Toxicol. 2006, 19, 577–586. [Google Scholar] [CrossRef]
- Gallagher, E.P.; Kunze, K.L.; Stapleton, P.L.; Eaton, D.L. The kinetics of aflatoxin B1 oxidation by human cDNA-expressed and human liver microsomal cytochromes P450 1A2 and 3A4. Toxicol. Appl. Pharmacol. 1996, 141, 595–606. [Google Scholar] [CrossRef]
- Van Vleet, T.R.; Mace, K.; Coulombe, R.A., Jr. Comparative aflatoxin B1 activation and cytotoxicity in human bronchial cells expressing cytochromes P450 1A2 and 3A4. Cancer Res. 2002, 62, 105–112. [Google Scholar]
- Kim, D.; Guengerich, F.P. Enhancement of 7-methoxyresorufin O-demethylation activity of human cytochrome P450 1A2 by molecular breeding. Arch. Biochem. Biophys. 2004, 432, 102–108. [Google Scholar] [CrossRef]
- Tu, Y.; Deshmukh, R.; Sivaneri, M.; Szklarz, G.D. Application of molecular modeling for prediction of substrate specificity in cytochrome P450 1A2 mutants. Drug Metab. Dispos. 2008, 36, 2371–2380. [Google Scholar] [CrossRef]
- Liu, J.; Ericksen, S.S.; Sivaneri, M.; Besspiata, D.; Fisher, C.W.; Szklarz, G.D. The effect of reciprocal active site mutations in human cytochromes P450 1A1 and 1A2 on alkoxyresorufin metabolism. Arch. Biochem. Biophys. 2004, 424, 33–43. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef]
- Shimada, T.; Tanaka, K.; Takenaka, S.; Murayama, N.; Martin, M.V.; Foroozesh, M.K.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Structure-function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem. Res. Toxicol. 2010, 23, 1921–1935. [Google Scholar] [CrossRef]
- Liu, J.; Nguyen, T.T.; Dupart, P.S.; Sridhar, J.; Zhang, X.; Zhu, N.; Stevens, C.L.; Foroozesh, M. 7-ethynylcoumarins: Selective inhibitors of human cytochrome P450s 1A1 and 1A2. Chem. Res. Toxicol. 2012, 25, 1047–1057. [Google Scholar] [CrossRef]
- Bonomo, S.; Jorgensen, F.S.; Olsen, L. Dissecting the cytochrome P450 1A2- and 3A4-mediated metabolism of aflatoxin B1 in ligand and protein contributions. Chemistry 2017, 23, 2884–2893. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL molecular graphics system, version 1.8. 2015. [Google Scholar]
- Holzwarth, G.; Doty, P. The ultraviolet circular dichroism of polypeptides. J. Am. Chem. Soc. 1965, 87, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, R.; Zhang, C.; Li, K.; Xu, W.; Wang, L.; Chen, Q.; Mu, P.; Jiang, J.; Wen, J.; et al. Bioactivation and regioselectivity of pig cytochrome P450 3A29 towards aflatoxin B1. Toxins 2016, 8, 267. [Google Scholar] [CrossRef]
- Parikh, A.; Josephy, P.D.; Guengerich, F.P. Selection and characterization of human cytochrome P450 1A2 mutants with altered catalytic properties. Biochemistry 1999, 38, 5283–5289. [Google Scholar] [CrossRef]
- Nishio, M.; Hirota, M.; Umezawa, Y. The CH/π interaction. Evidence, nature, and consequences. John Wiley & Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Fujii, A. Magnitude and directionality of the interaction energy of the aliphatic CH/π interaction: Significant difference from hydrogen bond. J. Phys. Chem. A 2006, 110, 10163–10168. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. The magnitude of the CH/π interaction between benzene and some model hydrocarbons. J. Am. Chem. Soc. 2000, 122, 3746–3753. [Google Scholar] [CrossRef]
- Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Chakrabarti, P. CH-π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef]
- Santana, A.G.; Jimenez-Moreno, E.; Gomez, A.M.; Corzana, F.; Gonzalez, C.; Jimenez-Oses, G.; Jimenez-Barbero, J.; Asensio, J.L. A dynamic combinatorial approach for the analysis of weak carbohydrate/aromatic complexes: Dissecting facial selectivity in CH/π stacking interactions. J. Am. Chem. Soc. 2013, 135, 3347–3350. [Google Scholar] [CrossRef]
- Ozawa, T.; Okazaki, K.; Kitaura, K. Importance of CH/π hydrogen bonds in recognition of the core motif in proline-recognition domains: An ab initio fragment molecular orbital study. J. Comput. Chem. 2011, 32, 2774–2782. [Google Scholar] [CrossRef]
- Ozawa, T.; Okazaki, K.; Kitaura, K. CH/π hydrogen bonds play a role in ligand recognition and equilibrium between active and inactive states of the β2 adrenergic receptor: An ab initio fragment molecular orbital (FMO) study. Bioorg. Med. Chem. 2011, 19, 5231–5237. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Okazaki, K. CH/π hydrogen bonds determine the selectivity of the Src homology 2 domain to tyrosine phosphotyrosyl peptides: An ab initio fragment molecular orbital study. J. Comput. Chem. 2008, 29, 2656–2666. [Google Scholar] [CrossRef]
- Harigai, M.; Kataoka, M.; Imamoto, Y. A single CH/π weak hydrogen bond governs stability and the photocycle of the photoactive yellow protein. J. Am. Chem. Soc. 2006, 128, 10646–10647. [Google Scholar] [CrossRef]
- Umezawa, Y.; Nishio, M. CH/π hydrogen bonds as evidenced in the substrate specificity of acetylcholine esterase. Biopolymers 2005, 79, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Brandl, M.; Weiss, M.S.; Jabs, A.; Suhnel, J.; Hilgenfeld, R. C-H…π-interactions in proteins. J. Mol. Biol. 2001, 307, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Davis, A.M.; Chohan, K.K.; Paine, S.W.; Boyer, S.; Gavaghan, C.L.; Arnby, C.H.; Kankkonen, C.; Albertson, N. Generation of in-silico cytochrome P450 1A2, 2C9, 2C19, 2D6, and 3A4 inhibition qsar models. J. Comput. Aided. Mol. Des. 2007, 21, 559–573. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water; AIP: West Lafayette, IN, USA, 1983; Volume 79, pp. 926–935. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Zhou, R.H.; Harder, E.; Xu, H.F.; Berne, B.J. Efficient multiple time step method for use with ewald and particle mesh ewald for large biomolecular systems. J. Chem. Phy. 2001, 115, 2348–2358. [Google Scholar] [CrossRef]
- Sandhu, P.; Guo, Z.; Baba, T.; Martin, M.V.; Tukey, R.H.; Guengerich, F.P. Expression of modified human cytochrome P450 1A2 in Escherichia coli: Stabilization, purification, spectral characterization, and catalytic activities of the enzyme. Arch. Biochem. Biophys. 1994, 309, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Phillips, I.R.; Shephard, E.A.; Montellano, P.R.O.D. Cytochrome P450 Protocols; Humana Press: London, UK, 2013. [Google Scholar]
- Miller, G.P.; Guengerich, F.P. Binding and oxidation of alkyl 4-nitrophenyl ethers by rabbit cytochrome P450 1A2: Evidence for two binding sites. Biochemistry 2001, 40, 7262–7272. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, W.; Zhang, C.; Chang, Q.; Tang, X.; Li, K.; Deng, Y. Trp266 determines the binding specificity of a porcine aflatoxin B1 aldehyde reductase for aflatoxin B1-dialdehyde. Biochem. Pharmacol. 2013, 86, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.D.; Eaton, D.L.; Guengerich, F.P.; Coulombe, R.A., Jr. Aflatoxin B1 activation in human lung. Toxicol. Appl. Pharmacol. 1997, 144, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Wang-Buhler, J.L.; Lee, S.J.; Chung, W.G.; Stevens, J.F.; Tseng, H.P.; Hseu, T.H.; Hu, C.H.; Westerfield, M.; Yang, Y.H.; Miranda, C.L.; et al. CYP 2K6 from zebrafish (Danio rerio): Cloning, mapping, developmental/tissue expression, and aflatoxin B1 activation by baculovirus expressed enzyme. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2005, 140, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Guengerich, F.P. Selection of human cytochrome P450 1A2 mutants with enhanced catalytic activity for heterocyclic amine n-hydroxylation. Biochemistry 2004, 43, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Miller, G.P.; Guengerich, F.P. Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry 2000, 39, 11319–11329. [Google Scholar] [CrossRef] [PubMed]
- Briggs, G.E.; Haldane, J.B. A note on the kinetics of enzyme action. Biochem. J. 1925, 19, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, L.; Menten, M.L. Die kinetik der invertinwirkung. Biochem. Z. 1913, 49, 333–369. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Proteins | Km (μM) | Kcat (min−1) | Kcat/Km (min−1·mM−1) | Normalized Values (Kcat/Km) |
|---|---|---|---|---|
| WT | 29.86 ± 2.17 | 0.24 ± 0.007 | 8.0 | 1 |
| T124A | 6.79 ± 0.79 | 0.20 ± 0.004 | 29.5 | 3.69 |
| F125A | ND | ND | ND | ND |
| F226A | ND | ND | ND | ND |
| F260A | 457.35 ± 387.66 | 0.096 ± 0.067 | 0.2 | 0.025 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Zhu, S.; Wu, Y.; Jiang, T.; Wang, L.; Jiang, J.; Wen, J.; Deng, Y. Multiple CH/π Interactions Maintain the Binding of Aflatoxin B1 in the Active Cavity of Human Cytochrome P450 1A2. Toxins 2019, 11, 158. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030158
Wu J, Zhu S, Wu Y, Jiang T, Wang L, Jiang J, Wen J, Deng Y. Multiple CH/π Interactions Maintain the Binding of Aflatoxin B1 in the Active Cavity of Human Cytochrome P450 1A2. Toxins. 2019; 11(3):158. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030158
Chicago/Turabian StyleWu, Jun, Sisi Zhu, Yunbo Wu, Tianqing Jiang, Lingling Wang, Jun Jiang, Jikai Wen, and Yiqun Deng. 2019. "Multiple CH/π Interactions Maintain the Binding of Aflatoxin B1 in the Active Cavity of Human Cytochrome P450 1A2" Toxins 11, no. 3: 158. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030158