Exploring the Role of Staphylococcus Aureus Toxins in Atopic Dermatitis

 , , , , and
, , , , and 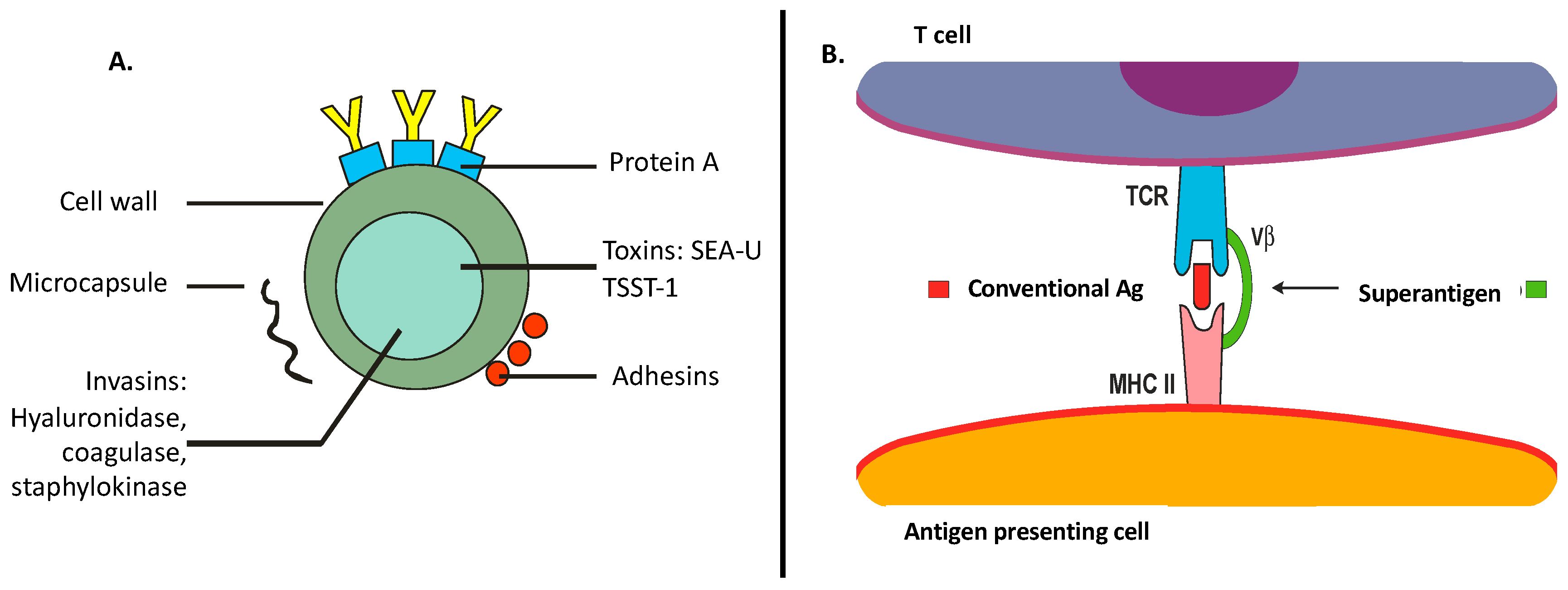{kind=link}
{kind=link}
Abstract
:Introduction
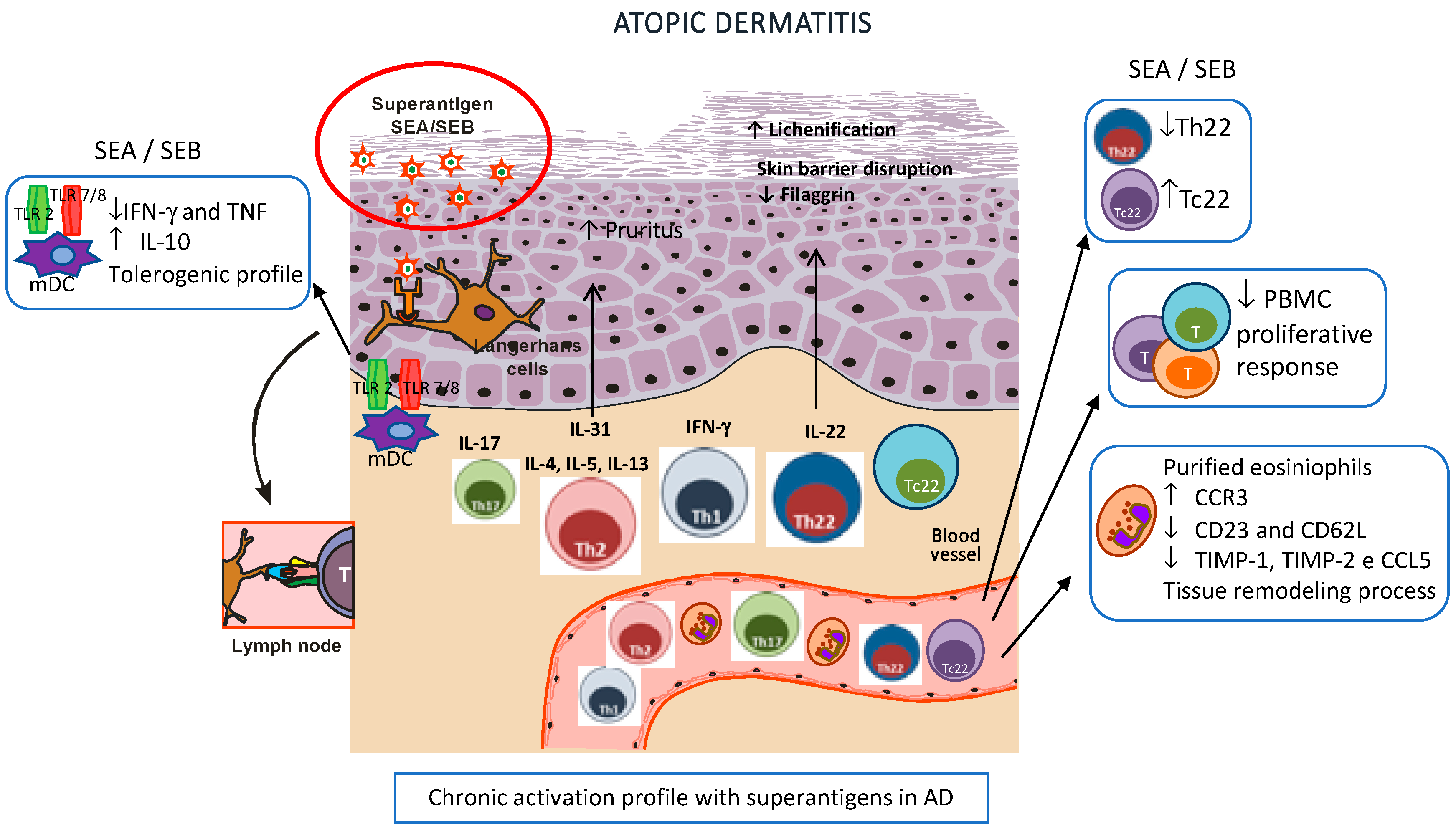
1. Role of S. Aureus Toxins in the Disruption of the Skin Barrier and Innate Immunity
2. S. Aureus: Straight Relationship with Adaptive Immunity in AD
3. Microbiome and AD
4. Perspectives Targeting S. Aureus in AD
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leung, D.Y.M. New insights into atopic dermatitis: Role of skin barrier and immune dysregulation. Allergol. Int. 2013, 62, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Schmuth, M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salava, A.; Lauerma, A. Role of the skin microbiome in atopic dermatitis. Clin. Trans. Allergy 2014, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, S.; Uchi, H.; Moroi, Y.; Furue, M. Decrease in circulating th17 cells correlates with increased levels of ccl17, ige and eosinophils in atopic dermatitis. J. Dermatol. Sci. 2011, 61, 180–186. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Agnihothri, R.; McGirt, L.Y.; Bankova, L.G.; Beck, L.A. Atopic dermatitis: A disease caused by innate immune defects? J. Investig. Dermatol. 2009, 129, 14–30. [Google Scholar] [CrossRef]
- Saeed, K.; Marsh, P.; Ahmad, N. Cryptic resistance in staphylococcus aureus: A risk for the treatment of skin infection? Curr. Opin. Infectious Dis. 2014, 27, 130–136. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Krakauer, T. Update on staphylococcal superantigen-induced signaling pathways and therapeutic interventions. Toxins 2013, 5, 1629–1654. [Google Scholar] [CrossRef]
- Paller, A.S.; Kong, H.H.; Seed, P.; Naik, S.; Scharschmidt, T.C.; Gallo, R.L.; Luger, T.; Irvine, A.D. The microbiome in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 26–35. [Google Scholar] [CrossRef]
- Oliveira, D.; Borges, A.; Simoes, M. Staphylococcus aureus toxins and their molecular activity in infectious diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.; Torres, V.J. Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol. Spectr. 2019, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Orfali, R.L.; Sato, M.N.; Takaoka, R.; Azor, M.H.; Rivitti, E.A.; Hanifin, J.M.; Aoki, V. Atopic dermatitis in adults: Evaluation of peripheral blood mononuclear cells proliferation response to staphylococcus aureus enterotoxins a and b and analysis of interleukin-18 secretion. Exp. Dermatology 2009, 18, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Tuffs, S.W.; Haeryfar, S.M.M.; McCormick, J.K. Manipulation of innate and adaptive immunity by staphylococcal superantigens. Pathogens 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A. Skin immunity. Arch. Immunol. Ther. Exp. (Warsz) 2018, 66, 45–54. [Google Scholar] [CrossRef]
- Cole, G.W.; Silverberg, N.L. The adherence of staphylococcus aureus to human corneocytes. Arch. Dermatol. 1986, 122, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Ezepchuk, Y.V.; Leung, D.Y.; Middleton, M.H.; Bina, P.; Reiser, R.; Norris, D.A. Staphylococcal toxins and protein a differentially induce cytotoxicity and release of tumor necrosis factor-alpha from human keratinocytes. J. Investig. Dermatol. 1996, 107, 603–609. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Niebuhr, M.; Heratizadeh, A.; Wichmann, K.; Satzger, I.; Werfel, T. Intrinsic alterations of pro-inflammatory mediators in unstimulated and tlr-2 stimulated keratinocytes from atopic dermatitis patients. Exp. Dermatol. 2011, 20, 468–472. [Google Scholar]
- Kissner, T.L.; Ruthel, G.; Cisney, E.D.; Ulrich, R.G.; Fernandez, S.; Saikh, K.U. Myd88-dependent pro-inflammatory cytokine response contributes to lethal toxicity of staphylococcal enterotoxin b in mice. Innate Immun. 2011, 17, 451–462. [Google Scholar] [CrossRef]
- Fassbender, S.; Opitz, F.V.; Johnen, S.; Forster, I.; Weighardt, H. Myd88 contributes to staphylococcal enterotoxin b-triggered atopic dermatitis-like skin inflammation in mice. J. Investig. Dermatol. 2017, 137, 1802–1804. [Google Scholar] [PubMed]
- Munoz-Planillo, R.; Franchi, L.; Miller, L.S.; Nunez, G. A critical role for hemolysins and bacterial lipoproteins in staphylococcus aureus-induced activation of the nlrp3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [PubMed]
- Niebuhr, M.; Baumert, K.; Heratizadeh, A.; Satzger, I.; Werfel, T. Impaired nlrp3 inflammasome expression and function in atopic dermatitis due to th2 milieu. Allergy 2014, 69, 1058–1067. [Google Scholar] [PubMed]
- Syed, A.K.; Reed, T.J.; Clark, K.L.; Boles, B.R.; Kahlenberg, J.M. Staphlyococcus aureus phenol-soluble modulins stimulate the release of proinflammatory cytokines from keratinocytes and are required for induction of skin inflammation. Infect. Immun. 2015, 83, 3428–3437. [Google Scholar] [PubMed]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Th2 cytokines increase staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (stat6). J. Investig. Dermatol. 2014, 134, 2114–2121. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Bin, L.; Kim, B.E.; Oyoshi, M.K.; Geha, R.S.; Goleva, E.; Leung, D.Y. Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal alpha-toxin-induced keratinocyte death. J. Allergy Clin. Immunol. 2013, 131, 421–427. [Google Scholar] [PubMed]
- Batista, D.I.S.; Perez, L.; Orfali, R.L.; Zaniboni, M.C.; Samorano, L.P.; Pereira, N.V.; Sotto, M.N.; Ishizaki, A.S.; Oliveira, L.M.S.; Sato, M.N.; et al. Profile of skin barrier proteins (filaggrin, claudins 1 and 4) and th1/th2/th17 cytokines in adults with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1091–1095. [Google Scholar] [CrossRef]
- Triplett, K.D.; Pokhrel, S.; Castleman, M.J.; Daly, S.M.; Elmore, B.O.; Joyner, J.A.; Sharma, G.; Herbert, G.; Campen, M.J.; Hathaway, H.J.; et al. Gper activation protects against epithelial barrier disruption by staphylococcus aureus alpha-toxin. Sci. Rep. 2019, 9, 1343. [Google Scholar]
- Brauweiler, A.M.; Hall, C.F.; Goleva, E.; Leung, D.Y.M. Staphylococcus aureus lipoteichoic acid inhibits keratinocyte differentiation through a p63-mediated pathway. J. Investig. Dermatol. 2017, 137, 2030–2033. [Google Scholar]
- Liu, H.; Archer, N.K.; Dillen, C.A.; Wang, Y.; Ashbaugh, A.G.; Ortines, R.V.; Kao, T.; Lee, S.K.; Cai, S.S.; Miller, R.J.; et al. Staphylococcus aureus epicutaneous exposure drives skin inflammation via il-36-mediated t cell responses. Cell Host Microbe. 2017, 22, 653–666 e655. [Google Scholar]
- Baldry, M.; Nakamura, Y.; Nakagawa, S.; Frees, D.; Matsue, H.; Nunez, G.; Ingmer, H. Application of an agr-specific antivirulence compound as therapy for staphylococcus aureus-induced inflammatory skin disease. J. Infect. Dis. 2018, 218, 1009–1013. [Google Scholar]
- Matsuo, K.; Nagakubo, D.; Komori, Y.; Fujisato, S.; Takeda, N.; Kitamatsu, M.; Nishiwaki, K.; Quan, Y.S.; Kamiyama, F.; Oiso, N.; et al. Ccr4 is critically involved in skin allergic inflammation of balb/c mice. J. Investig. Dermatol. 2018, 138, 1764–1773. [Google Scholar] [PubMed]
- Kim, B.S.; Choi, J.K.; Jung, H.J.; Park, K.H.; Jang, Y.H.; Lee, W.J.; Lee, S.J.; Kim, S.H.; Kang, H.Y.; Kim, J.M.; et al. Effects of topical application of a recombinant staphylococcal enterotoxin a on dncb and dust mite extract-induced atopic dermatitis-like lesions in a murine model. Eur. J. Dermatol. 2014, 24, 186–193. [Google Scholar]
- Lee, H.W.; Kim, S.M.; Kim, J.M.; Oh, B.M.; Kim, J.Y.; Jung, H.J.; Lim, H.J.; Kim, B.S.; Lee, W.J.; Lee, S.J.; et al. Potential immunoinflammatory role of staphylococcal enterotoxin a in atopic dermatitis: Immunohistopathological analysis and in vitro assay. Ann. Dermatol. 2013, 25, 173–180. [Google Scholar] [PubMed]
- Saloga, J.; Leung, D.Y.; Reardon, C.; Giorno, R.C.; Born, W.; Gelfand, E.W. Cutaneous exposure to the superantigen staphylococcal enterotoxin b elicits a t-cell-dependent inflammatory response. J. Investig. Dermatol. 1996, 106, 982–988. [Google Scholar] [PubMed]
- Azimi, E.; Reddy, V.B.; Lerner, E.A. Brief communication: Mrgprx2, atopic dermatitis and red man syndrome. Itch (Philadelphia, Pa.) 2017, 2, e5. [Google Scholar]
- Ackermann, L.; Pelkonen, J.; Harvima, I.T. Staphylococcal enterotoxin b inhibits the production of interleukin-4 in a human mast-cell line hmc-1. Immunology 1998, 94, 247–252. [Google Scholar]
- Jorgensen, J.; Bach-Mortensen, N.; Koch, C.; Fomsgaard, A.; Baek, L.; Jarlov, J.O.; Espersen, F.; Jensen, C.B.; Skov, P.S.; Norn, S. Bacteria and endotoxin induce release of basophil histamine in patients with atopic dermatitis. In vitro experiments with s. Aureus, teichoic acid, e. Coli and e. Coli lps. Allergy 1987, 42, 395–397. [Google Scholar]
- Wehner, J.; Neuber, K. Staphylococcus aureus enterotoxins induce histamine and leukotriene release in patients with atopic eczema. Br. J. Dermatol. 2001, 145, 302–305. [Google Scholar]
- Minai-Fleminger, Y.; Gangwar, R.S.; Migalovich-Sheikhet, H.; Seaf, M.; Leibovici, V.; Hollander, N.; Feld, M.; Moses, A.E.; Homey, B.; Levi-Schaffer, F. The cd48 receptor mediates staphylococcus aureus human and murine eosinophil activation. Clin. Exp. Allergy 2014, 44, 1335–1346. [Google Scholar]
- Wedi, B.; Wieczorek, D.; Stunkel, T.; Breuer, K.; Kapp, A. Staphylococcal exotoxins exert proinflammatory effects through inhibition of eosinophil apoptosis, increased surface antigen expression (cd11b, cd45, cd54, and cd69), and enhanced cytokine-activated oxidative burst, thereby triggering allergic inflammatory reactions. J. Allergy Clin. Immunol. 2002, 109, 477–484. [Google Scholar] [PubMed]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, V.G.; Orfali, R.L.; Titz, T.D.; Duarte, A.J.D.; Sato, M.N.; Aoki, V. Evidence of regulatory myeloid dendritic cells and circulating inflammatory epidermal dendritic cells-like modulated by toll-like receptors 2 and 7/8 in adults with atopic dermatitis. Int. J. Dermatol. 2017, 56, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Mandron, M.; Aries, M.F.; Brehm, R.D.; Tranter, H.S.; Acharya, K.R.; Charveron, M.; Davrinche, C. Human dendritic cells conditioned with staphylococcus aureus enterotoxin b promote th2 cell polarization. J. Allergy Clin. Immunol. 2006, 117, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Kasraie, S.; Niebuhr, M.; Werfel, T. Interleukin (il)-31 induces pro-inflammatory cytokines in human monocytes and macrophages following stimulation with staphylococcal exotoxins. Allergy 2010, 65, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Kapitany, A.; Beke, G.; Nagy, G.; Doan-Xuan, Q.M.; Bacso, Z.; Gaspar, K.; Boros, G.; Dajnoki, Z.; Biro, T.; Rajnavolgyi, E.; et al. Cd1c+ blood dendritic cells in atopic dermatitis are premature and can produce disease-specific chemokines. Acta Derm. -Venereol. 2017, 97, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Oh, M.H.; Park, J.U.; Myers, A.C.; Dong, C.; Zhu, Z.; Zheng, T. Epicutaneous exposure to staphylococcal superantigen enterotoxin b enhances allergic lung inflammation via an il-17a dependent mechanism. PLoS ONE 2012, 7, e39032. [Google Scholar]
- Bin, L.; Kim, B.E.; Brauweiler, A.; Goleva, E.; Streib, J.; Ji, Y.; Schlievert, P.M.; Leung, D.Y. Staphylococcus aureus alpha-toxin modulates skin host response to viral infection. J. Allergy Clin. Immunol. 2012, 130, 683–691 e682. [Google Scholar] [CrossRef]
- Zhang, X.; Shang, W.; Yuan, J.; Hu, Z.; Peng, H.; Zhu, J.; Hu, Q.; Yang, Y.; Liu, H.; Jiang, B.; et al. Positive feedback cycle of tnfalpha promotes staphylococcal enterotoxin b-induced thp-1 cell apoptosis. Front. Cell. Infection Microbiol. 2016, 6, 109. [Google Scholar]
- Kedzierska, A.; Kaszuba-Zwoinska, J.; Slodowska-Hajduk, Z.; Kapinska-Mrowiecka, M.; Czubak, M.; Thor, P.; Wojcik, K.; Pryjma, J. Seb-induced t cell apoptosis in atopic patients--correlation to clinical status and skin colonization by staphylococcus aureus. Arch. Immunol. Ther. Exp. 2005, 53, 63–70. [Google Scholar]
- Bratton, D.L.; May, K.R.; Kailey, J.M.; Doherty, D.E.; Leung, D.Y. Staphylococcal toxic shock syndrome toxin-1 inhibits monocyte apoptosis. J. Allergy Clin. Immunol. 1999, 103, 895–900. [Google Scholar] [CrossRef]
- Mandron, M.; Aries, M.F.; Boralevi, F.; Martin, H.; Charveron, M.; Taieb, A.; Davrinche, C. Age-related differences in sensitivity of peripheral blood monocytes to lipopolysaccharide and staphylococcus aureus toxin b in atopic dermatitis. J. Investig. Dermatol. 2008, 128, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Kasraie, S.; Niebuhr, M.; Kopfnagel, V.; Dittrich-Breiholz, O.; Kracht, M.; Werfel, T. Macrophages from patients with atopic dermatitis show a reduced cxcl10 expression in response to staphylococcal alpha-toxin. Allergy 2012, 67, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Lutat, C.; Sigel, S.; Werfel, T. Impaired tlr-2 expression and tlr-2-mediated cytokine secretion in macrophages from patients with atopic dermatitis. Allergy 2009, 64, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Schorling, K.; Heratizadeh, A.; Werfel, T. Staphylococcal alpha-toxin induces a functional upregulation of tlr-2 on human peripheral blood monocytes. Exp. Dermatol. 2015, 24, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Krogman, A.; Tilahun, A.; David, C.S.; Chowdhary, V.R.; Alexander, M.P.; Rajagopalan, G. Hla-dr polymorphisms influence in vivo responses to staphylococcal toxic shock syndrome toxin-1 in a transgenic mouse model. HLA 2017, 89, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Langnickel, J.; Draing, C.; Renz, H.; Kapp, A.; Werfel, T. Dysregulation of toll-like receptor-2 (tlr-2)-induced effects in monocytes from patients with atopic dermatitis: Impact of the tlr-2 r753q polymorphism. Allergy 2008, 63, 728–734. [Google Scholar] [CrossRef]
- Huang, K.; Ran, L.; Wang, W.; Zhou, R.; Cai, X.; Li, R.; Li, Y.; Zhou, C.; He, W.; Wang, R. Glucocorticoid insensitivity by staphylococcal enterotoxin b in keratinocytes of allergic dermatitis is associated with impaired nuclear translocation of the glucocorticoid receptor alpha. J. Dermatological Sci. 2018, 92, 272–280. [Google Scholar] [CrossRef]
- Stoll, H.; Ost, M.; Singh, A.; Mehling, R.; Neri, D.; Schafer, I.; Velic, A.; Macek, B.; Kretschmer, D.; Weidenmaier, C.; et al. Staphylococcal enterotoxins dose-dependently modulate the generation of myeloid-derived suppressor cells. Front. Cell. Infection Microbiol. 2018, 8, 321. [Google Scholar] [CrossRef]
- Wichmann, K.; Uter, W.; Weiss, J.; Breuer, K.; Heratizadeh, A.; Mai, U.; Werfel, T. Isolation of alpha-toxin-producing staphylococcus aureus from the skin of highly sensitized adult patients with severe atopic dermatitis. Br. J. Dermatol. 2009, 161, 300–305. [Google Scholar] [CrossRef]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; Program, N.C.S.; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesner, L.M.; Werfel, T.; Heratizadeh, A. The adaptive immune system in atopic dermatitis and implications on therapy. Expert. Rev. Clin. Immunol. 2016, 12, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Wittmann, M.; Kempe, K.; Kapp, A.; Mai, U.; Dittrich-Breiholz, O.; Kracht, M.; Mrabet-Dahbi, S.; Werfel, T. Alpha-toxin is produced by skin colonizing staphylococcus aureus and induces a t helper type 1 response in atopic dermatitis. Clin. Exp. Allergy 2005, 35, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, K.; Novak, N. Immunology of atopic eczema: Overcoming the th1/th2 paradigm. Allergy 2013, 68, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, T.; Skabytska, Y.; Kaesler, S.; Volz, T. Regulation of t cell immunity in atopic dermatitis by microbes: The yin and yang of cutaneous inflammation. Front. Immunol. 2015, 6, 353. [Google Scholar] [CrossRef]
- Auriemma, M.; Vianale, G.; Amerio, P.; Reale, M. Cytokines and t cells in atopic dermatitis. Eur. Cytokine Netw. 2013, 24, 37–44. [Google Scholar]
- Koga, C.; Kabashima, K.; Shiraishi, N.; Kobayashi, M.; Tokura, Y. Possible pathogenic role of th17 cells for atopic dermatitis. J. Investig. Dermatol. 2008, 128, 2625–2630. [Google Scholar] [CrossRef]
- D’Auria, E.; Banderali, G.; Barberi, S.; Gualandri, L.; Pietra, B.; Riva, E.; Cerri, A. Atopic dermatitis: Recent insight on pathogenesis and novel therapeutic target. Asian-Pac. J. Allergy Immunol. 2016, 34, 98–108. [Google Scholar]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (il)-22 and il-17 are coexpressed by th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory t cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef]
- Orfali, R.L.; da Silva Oliveira, L.M.; de Lima, J.F.; de Carvalho, G.C.; Ramos, Y.A.L.; Pereira, N.Z.; Pereira, N.V.; Zaniboni, M.C.; Sotto, M.N.; da Silva Duarte, A.J.; et al. Staphylococcus aureus enterotoxins modulate il-22-secreting cells in adults with atopic dermatitis. Sci. Rep. 2018, 8, 6665. [Google Scholar] [CrossRef] [PubMed]
- Laborel-Preneron, E.; Bianchi, P.; Boralevi, F.; Lehours, P.; Fraysse, F.; Morice-Picard, F.; Sugai, M.; Sato’o, Y.; Badiou, C.; Lina, G.; et al. Effects of the staphylococcus aureus and staphylococcus epidermidis secretomes isolated from the skin microbiota of atopic children on cd4+ t cell activation. PLoS ONE 2015, 10, e0141067. [Google Scholar]
- Simpson, E.L.; Villarreal, M.; Jepson, B.; Rafaels, N.; David, G.; Hanifin, J.; Taylor, P.; Boguniewicz, M.; Yoshida, T.; De Benedetto, A.; et al. Patients with atopic dermatitis colonized with staphylococcus aureus have a distinct phenotype and endotype. J. Investig. Dermatol. 2018, 138, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Titz, T.D.; Orfali, R.L.; de Lollo, C.; dos Santos, V.G.; Duarte, A.J.D.; Sato, M.N.; Aoki, V. Impaired cd23 and cd62l expression and tissue inhibitors of metalloproteinases secretion by eosinophils in adults with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 2072–2076. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Yang, Y.H.; Hwang, Y.W.; Tsai, M.J.; Tsao, P.N.; Chiang, B.L.; Shau, W.Y.; Wang, L.F. Comparison of serum specific ige antibodies to staphylococcal enterotoxins between atopic children with and without atopic dermatitis. Allergy 2000, 55, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Suzuki, S.; Ohta, S.; Manabe, R.; Furukawa, H.; Kuwahara, N.; Fukuda, Y.; Kimura, T.; Jinno, M.; Hirai, K.; et al. Association between specific ige to staphylococcus aureus enterotoxins a and b and asthma control. Ann. Allerg Asthma Immunol. 2015, 115, 191. [Google Scholar] [CrossRef] [PubMed]
- Parcina, M.; Miranda-Garcia, M.A.; Durlanik, S.; Ziegler, S.; Over, B.; Georg, P.; Foermer, S.; Ammann, S.; Hilmi, D.; Weber, K.J.; et al. Pathogen-triggered activation of plasmacytoid dendritic cells induces il-10-producing b cells in response to staphylococcus aureus. J. Immunol. 2013, 190, 1591–1602. [Google Scholar] [CrossRef]
- Boccardi, D.; D’Auria, E.; Turati, F.; DI, M.V.; Sortino, S.; Riva, E.; Cerri, A. Disease severity and quality of life in children with atopic dermatitis: Po-scorad in clinical practice. Minerva Pediatrica 2017, 69, 373–380. [Google Scholar]
- Coutanceau, C.; Stalder, J.F. Analysis of correlations between patient-oriented scorad (po-scorad) and other assessment scores of atopic dermatitis severity and quality of life. Dermatology 2014, 229, 248–255. [Google Scholar] [CrossRef]
- Raap, U.; Wichmann, K.; Bruder, M.; Stander, S.; Wedi, B.; Kapp, A.; Werfel, T. Correlation of il-31 serum levels with severity of atopic dermatitis. J. Allergy Clin. Immunol. 2008, 122, 421–423. [Google Scholar] [CrossRef]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.C.; Meller, S.; Rieker, J.; et al. Il-31: A new link between t cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Werfel, T. Innate immunity, allergy and atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [Green Version]
- Kuo, I.H.; Yoshida, T.; De Benedetto, A.; Beck, L.A. The cutaneous innate immune response in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2013, 131, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.; Allam, J.P.; Biedermann, T.; Eyerich, K.; Gilles, S.; Guttman-Yassky, E.; Hoetzenecker, W.; Knol, E.; Simon, H.U.; Wollenberg, A.; et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 336–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J. Investig. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef]
- Schommer, N.N.; Gallo, R.L. Structure and function of the human skin microbiome. Trends Microbiol. 2013, 21, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.A.; Connolly, J.; Hourihane, J.O.; Fallon, P.G.; McLean, W.H.; Murray, D.; Jo, J.H.; Segre, J.A.; Kong, H.H.; Irvine, A.D. Skin microbiome before development of atopic dermatitis: Early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2017, 139, 166–172. [Google Scholar] [CrossRef]
- Shi, B.; Bangayan, N.J.; Curd, E.; Taylor, P.A.; Gallo, R.L.; Leung, D.Y.M.; Li, H. The skin microbiome is different in pediatric versus adult atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 1233–1236. [Google Scholar] [Green Version]
- Parlet, C.P.; Brown, M.M.; Horswill, A.R. Commensal staphylococci influence staphylococcus aureus skin colonization and disease. Trends Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Clowry, J.; Irvine, A.D.; McLoughlin, R.M. Next-generation anti-staphylococcus aureus vaccines: A potential new therapeutic option for atopic dermatitis? J. Allergy Clin. Immunol. 2019, 143, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, T.; Hanifin, J.M.; Furue, M.; Pulka, G.; Mlynarczyk, I.; Wollenberg, A.; Galus, R.; Etoh, T.; Mihara, R.; Yoshida, H.; et al. Anti-interleukin-31 receptor a antibody for atopic dermatitis. New Engl. J. Med. 2017, 376, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Myles, I.A.; Earland, N.J.; Anderson, E.D.; Moore, I.N.; Kieh, M.D.; Williams, K.W.; Saleem, A.; Fontecilla, N.M.; Welch, P.A.; Darnell, D.A.; et al. First-in-human topical microbiome transplantation with roseomonas mucosa for atopic dermatitis. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Blicharz, L.; Rudnicka, L.; Samochocki, Z. Staphylococcus aureus: An underestimated factor in the pathogenesis of atopic dermatitis? Adv. Dermatol. Allergol. 2019, 36, 11–17. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seiti Yamada Yoshikawa, F.; Feitosa de Lima, J.; Notomi Sato, M.; Álefe Leuzzi Ramos, Y.; Aoki, V.; Leao Orfali, R. Exploring the Role of Staphylococcus Aureus Toxins in Atopic Dermatitis. Toxins 2019, 11, 321. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11060321
Seiti Yamada Yoshikawa F, Feitosa de Lima J, Notomi Sato M, Álefe Leuzzi Ramos Y, Aoki V, Leao Orfali R. Exploring the Role of Staphylococcus Aureus Toxins in Atopic Dermatitis. Toxins. 2019; 11(6):321. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11060321
Chicago/Turabian StyleSeiti Yamada Yoshikawa, Fabio, Josenilson Feitosa de Lima, Maria Notomi Sato, Yasmin Álefe Leuzzi Ramos, Valeria Aoki, and Raquel Leao Orfali. 2019. "Exploring the Role of Staphylococcus Aureus Toxins in Atopic Dermatitis" Toxins 11, no. 6: 321. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11060321