T-2 Toxin-Induced Oxidative Stress Leads to Imbalance of Mitochondrial Fission and Fusion to Activate Cellular Apoptosis in the Human Liver 7702 Cell Line



Abstract
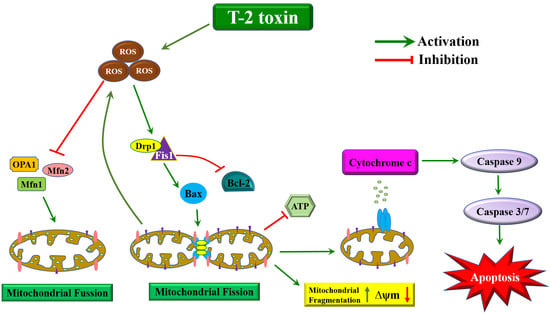
:
1. Introduction
2. Results
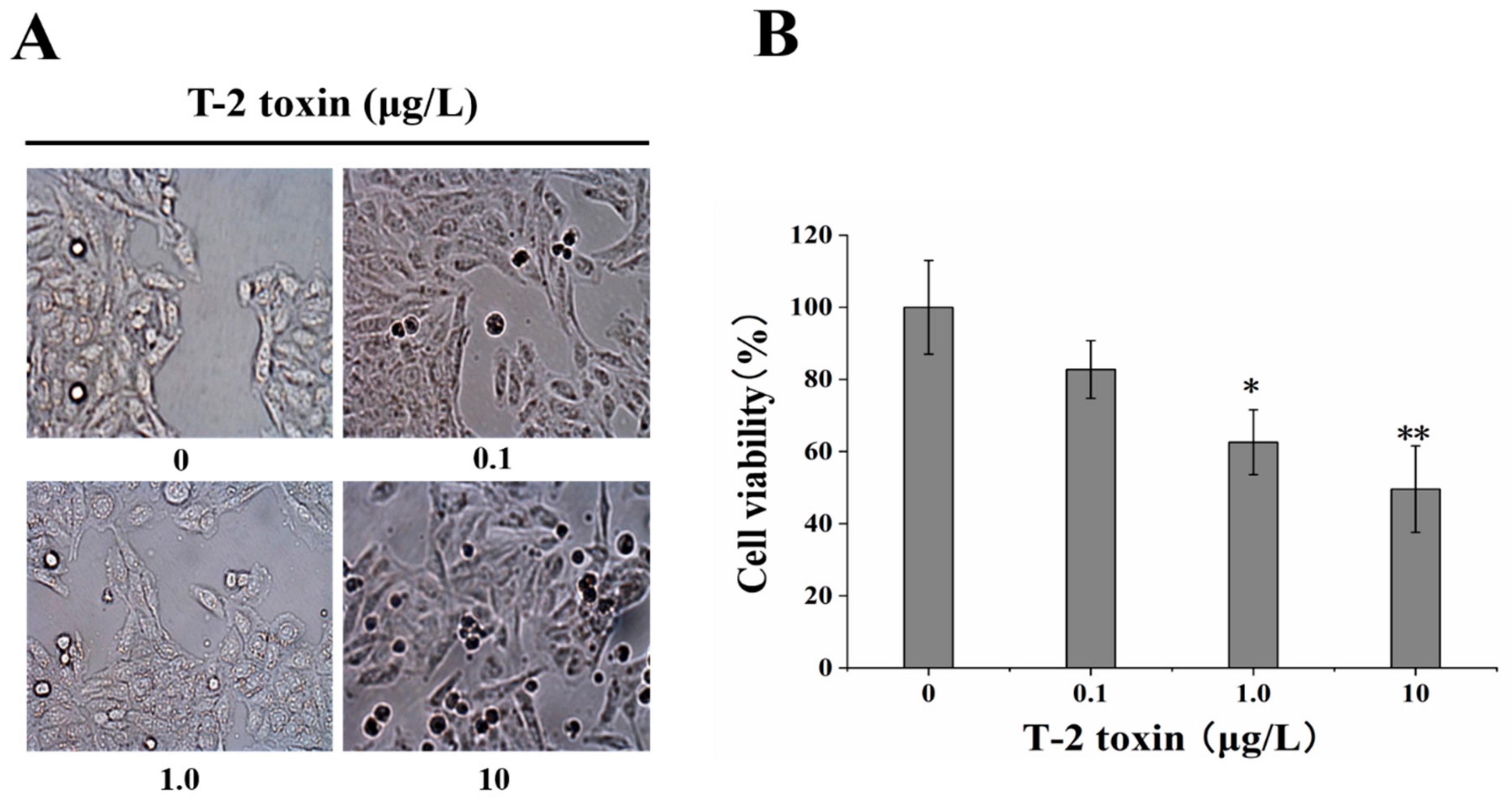
2.1. Effect of T-2 Toxin on Changes in Cell Morphology and Viability
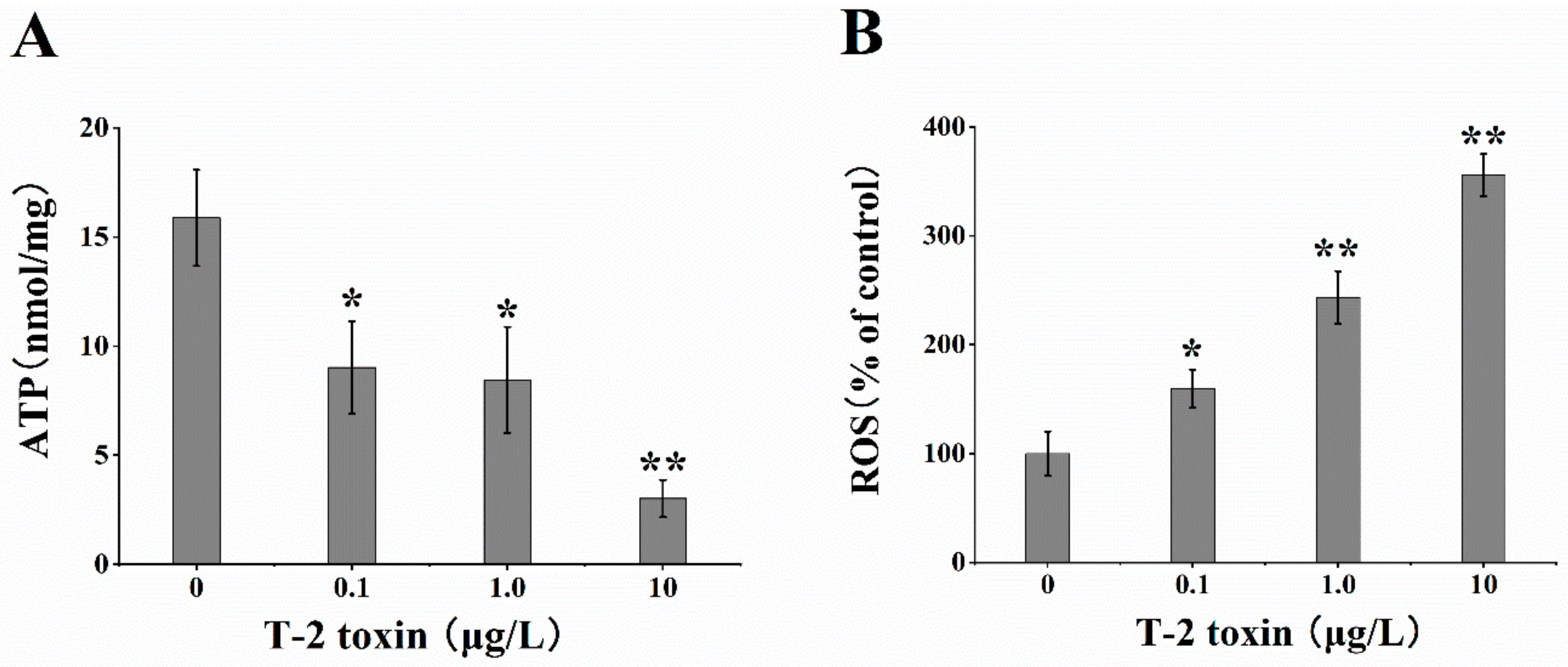
2.2. Effects of T-2 Toxin on the Generation of ATP and ROS
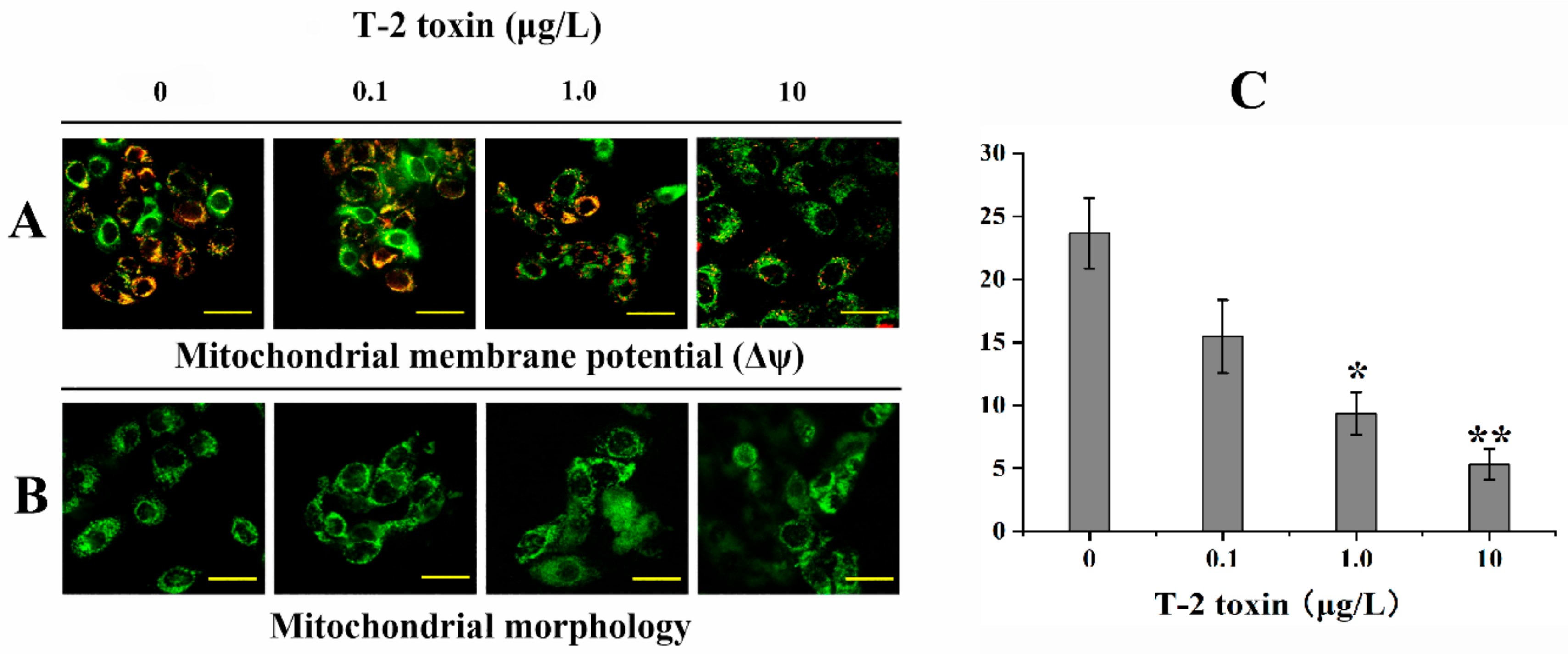
2.3. Effects of T-2 Toxin on Mitochondrial Function
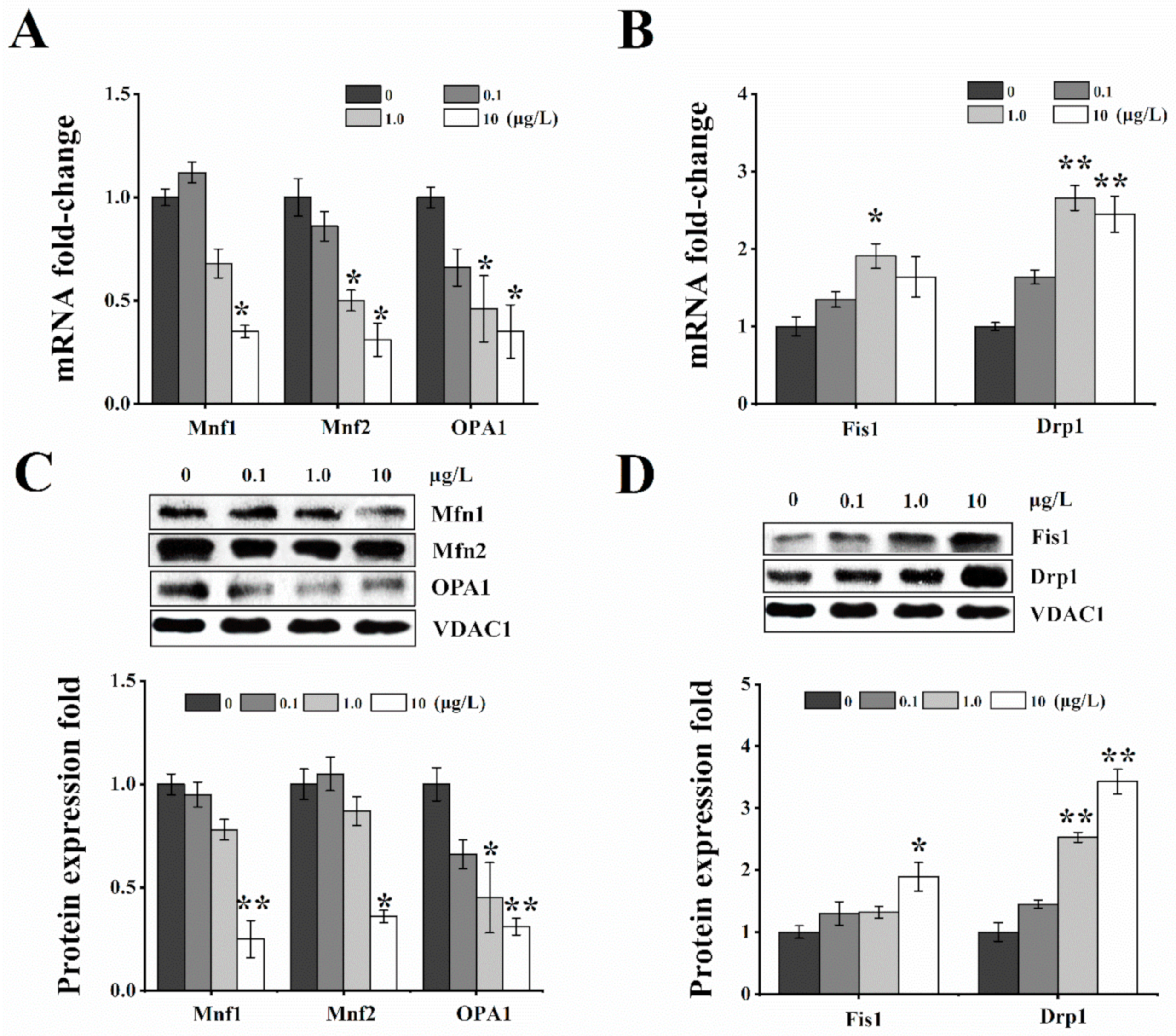
2.4. Effects of T-2 Toxin on Mitochondrial Dynamics-Related Genes and Proteins
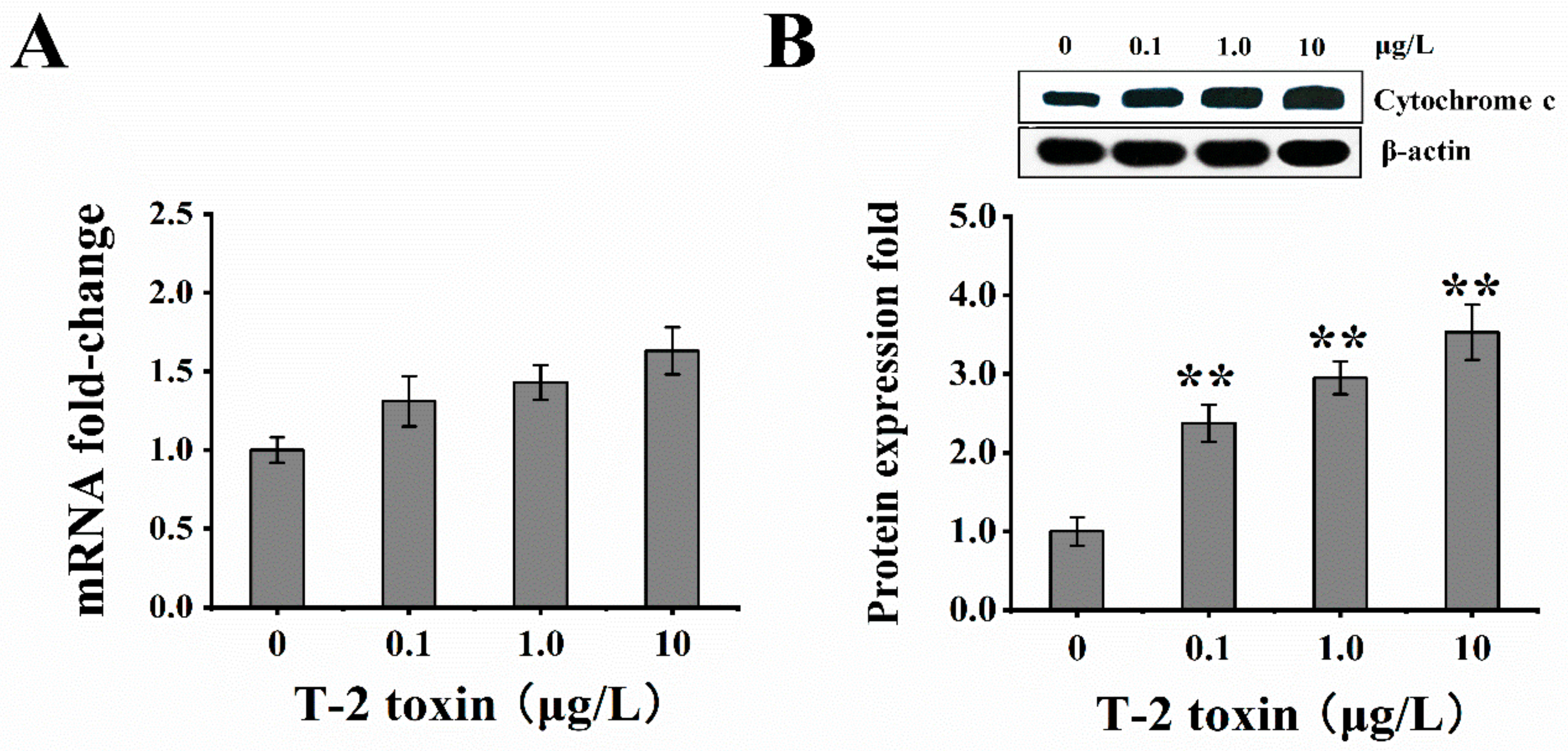
2.5. T-2 Toxin Induced Cytochrome C Release into the Cell
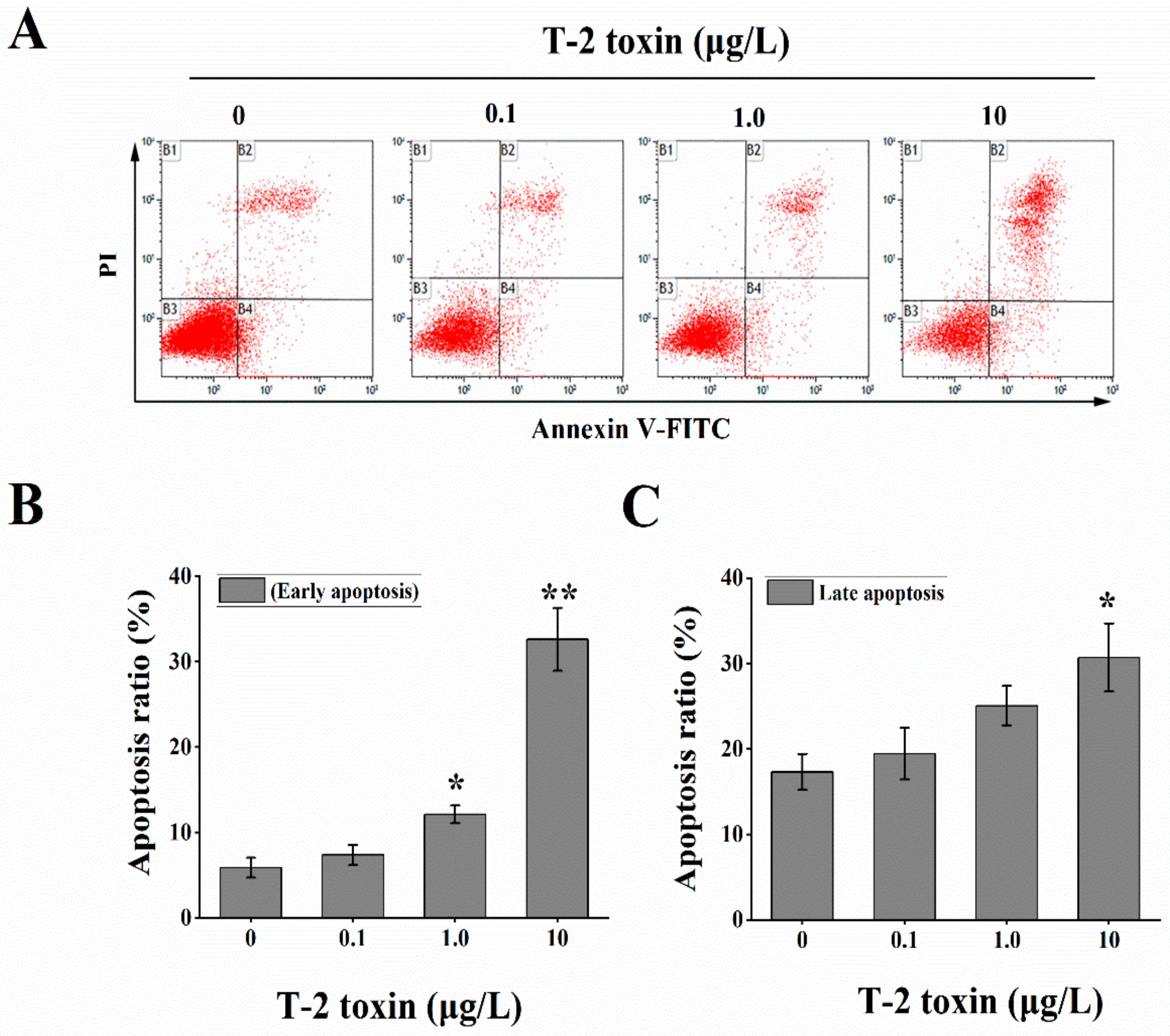
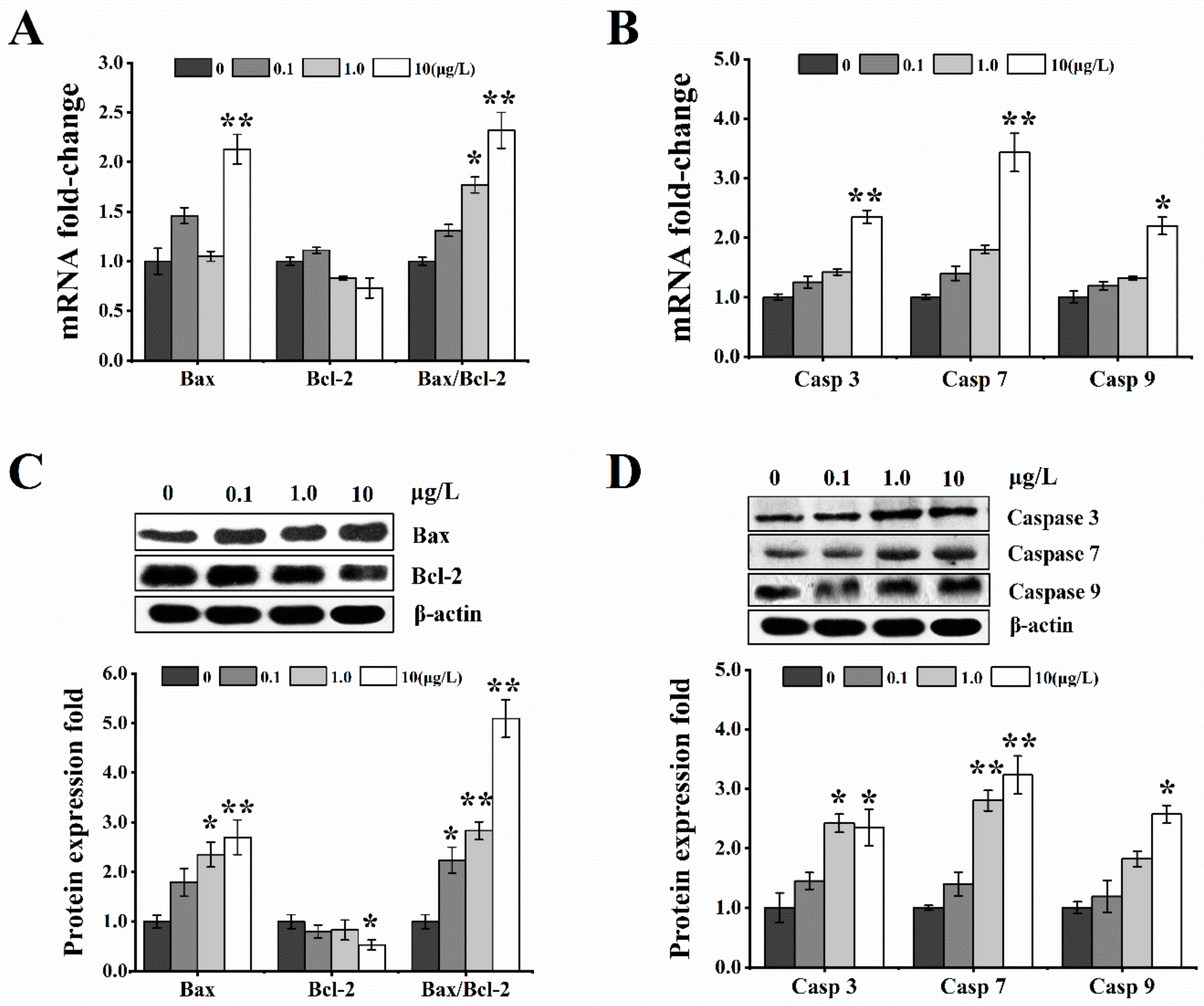
2.6. Effects of T-2 Toxin on Mitochondrial-Mediated Cellular Apoptosis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Measurement of Cell Viability
4.4. Measurement of Cellular ATP Level and ROS Generation
4.5. Measurement of Mitochondrial Membrane Potential (∆ψm) and Mitochondrial Morphology
4.6. Measurement of Cellular Apoptosis
4.7. RNA Extraction and Real-Time Quantitative PCR
4.8. Protein Extraction and Western Blotting Analysis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yu, F.F.; Lin, X.L.; Yang, L.; Liu, H.; Wang, X.; Fang, H.; Lammi, Z.J.; Guo, X. Comparison of T-2 toxin and HT-2 toxin distributed in the skeletal system with that in other tissues of rats by acute toxicity test. Biomed. Environ. Sci. 2017, 30, 851–854. [Google Scholar] [PubMed]
- Wang, J.P.; Duan, S.X.; Zhang, Y.; Wang, S. Enzyme-linked immunosorbent assay for the determination of T-2 toxin in cereals and feedstuff. Food Addit. Contam. Part B Surveill. 2016, 9, 275–280. [Google Scholar] [CrossRef]
- Wu, Q.; Dohnal, V.; Huang, L.; Kuca, K.; Yuan, Z. Metabolic pathways of trichothecenes. Drug Metab. Rev. 2010, 42, 250–267. [Google Scholar] [CrossRef]
- Makowska, K.; Gonkowski, S.; Zielonka, L.; Dabrowski, M.; Calka, J. T2 toxin-induced changes in cocaine- and amphetamine-regulated transcript (cart)-like immunoreactivity in the enteric nervous system within selected fragments of the porcine digestive tract. Neurotox. Res. 2017, 31, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Carlo, M.J.; Loeser, R.F. Nitric oxide-ediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002, 46, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhao, Z.J.; Ren, X.B.; Li, Q.; Ding, H.; Sun, Z.; Kao, Q.J.; Wang, L.H. Serum metabonomics of articular cartilage destruction induced by T-2 toxin in Wistar rats. Biomed. Environ. Sci. 2018, 31, 76–80. [Google Scholar]
- Wan, D.; Wang, X.; Wu, Q.; Lin, P.; Pan, Y.; Sattar, A.; Huang, L.; Ahmad, I.; Zhang, Y.; Yuan, Z. Integrated transcriptional and proteomic analysis of growth hormone suppression mediated by trichothecene t-2 toxin in rat gh3 cells. Toxicol. Sci. 2015, 147, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Q.; Ihsan, A.; Huang, L.; Dai, M.; Hao, H.; Cheng, G.; Liu, Z.; Wang, Y.; Yuan, Z. JAK/STAT pathway plays a critical role in the proinflammatory gene expression and apoptosis of RAW264.7 cells induced by trichothecenes as DON and T-2 toxin. Toxicol. Sci. 2012, 127, 412–424. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, D.; Huang, Y.; Wang, S. Study on the apoptosis mechanism induced by T-2 toxin. PLoS ONE 2013, 8, e83105. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, M.; Jayaraj, R.; Santhosh, S.R.; Rao, P.V. Oxidative damage and gene expression profile of antioxidant enzymes after T-2 toxin exposure in mice. J. Biochem. Mol. Toxicol. 2009, 23, 212–221. [Google Scholar] [CrossRef]
- Fang, H.; Wu, Y.; Guo, J.; Rong, J.; Ma, L.; Zhao, Z.; Zuo, D.; Peng, S. T-2 toxin induces apoptosis in differentiated murine embryonic stem cells through reactive oxygen species-mediated mitochondrial pathway. Apoptosis 2012, 17, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, M.; Bhaskar, A.S.; Rao, P.V. Differential effects of route of T-2 toxin exposure on hepatic oxidative damage in mice. Environ. Toxicol. 2015, 30, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Guo, X.; Pang, Q.; Wu, S.; Wu, C.; Xu, P.; Bai, Y. The role of mitochondria in T-2 toxin-induced human chondrocytes apoptosis. PLoS ONE 2014, 29, e108394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavi, M.; Rezaei, M.; Kalantari, H.; Behfar, A.; Varnaseri, G. L-carnitine protects rat hepatocytes from oxidative stress induced by T-2 toxin. Drug Chem. Toxicol. 2016, 39, 445–450. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Aradhyam, G.K. Identification and characterization of signal peptide of Mitofusin1 (Mfn1). Biochem. Biophys. Res. Commun. 2019, 509, 707–712. [Google Scholar] [CrossRef]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhao, X.; Ni, C.; Dai, Y.; Guo, Y. Zearalenone regulates endometrial stromal cell apoptosis and migration via the promotion of mitochondrial fission by activation of the JNK/Drp1 pathway. Mol. Med. Rep. 2018, 17, 7797–7806. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Dong, Z.; Lian, W.; Cui, J.; Wang, J.; Shen, H.; Liu, W.; Yang, J.; Zhang, X.; Cui, H. Ochratoxin A causes mitochondrial dysfunction, apoptotic and autophagic cell death and also induces mitochondrial biogenesis in human gastric epithelium cells. Arch. Toxicol. 2019, 93, 1141–1155. [Google Scholar] [CrossRef]
- Wang, W.; Wang, R.; Zhang, Q.; Mor, G.; Zhang, H. Benzo(a)pyren-7,8-dihydrodiol-9,10-epoxide induces human trophoblast Swan 71 cell dysfunctions due to cell apoptosis through disorder of mitochondrial fission/fusion. Environ. Pollut. 2018, 233, 820–832. [Google Scholar] [CrossRef]
- Jin, X.; Xu, Z.; Zhao, X.; Chen, M.; Xu, S. The antagonistic effect of selenium on lead-induced apoptosis via mitochondrial dynamics pathway in the chicken kidney. Chemosphere 2017, 180, 259–266. [Google Scholar] [CrossRef]
- Shao, Y.; Zhao, H.; Wang, Y.; Liu, J.; Zong, H.; Xing, M. Copper-mediated mitochondrial fission/fusion is associated with intrinsic apoptosis and autophagy in the testis tissues of chicken. Biol. Trace Elem. Res. 2019, 188, 468–477. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Zhang, J.; Ji, Q.; Huang, W.; Zhang, X.; Li, Y. Spermatogenesis disorder caused by T-2 toxin is associated with germ cell apoptosis mediated by oxidative stress. Environ. Pollut. 2019, 251, 372–379. [Google Scholar] [CrossRef]
- Ngampongsa, S.; Hanafusa, M.; Ando, K.; Ito, K.; Kuwahara, M.; Yamamoto, Y.; Yamashita, M.; Tsuru, Y.; Tsubone, H. Toxic effects of T-2 toxin and deoxynivalenol on the mitochondrial electron transport system of cardiomyocytes in rats. J. Toxicol. Sci. 2013, 38, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hymery, N.; Sibiril, Y.; Parent-Massin, D. In vitro effects of trichothecenes on human dendritic cells. Toxicol. In Vitro 2006, 20, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, C.; Bouslimi, A.; Kadri, R.; Zaied, C.; Bacha, H.; Abid-Essefi, S. The in vitro effects of zearalenone and T-2 toxins on Vero cells. Exp. Toxicol. Pathol. 2013, 65, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Hoftus, J.H.; Kijanka, G.S.; O’Kennedy, R.; Loscher, C.E. Patulin, Deoxynivalenol, Zearalenone and T-2 Toxin Affect Viability and Modulate Cytokine Secretion in J774A.1 Murine Macrophages. Int. J. Chem. 2016, 8, 22–32. [Google Scholar]
- Karacaoğlu, E.; Selmanoğlu, G. T-2 toxin induces cytotoxicity and disrupts tight junction barrier in SerW3 cells. Environ. Toxicol. Pharmacol. 2017, 56, 259–267. [Google Scholar] [CrossRef]
- Chaudhari, M.; Jayaraj, R.; Bhaskar, A.S.; Lakshmana Rao, P.V. Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology 2009, 262, 153–161. [Google Scholar] [CrossRef]
- Wu, J.; Huang, W.M.; Xiao, H.S.; Xie, Y.; Yuan, Z.H.; Yi, J.; Chen, J.S.; Tu, D.; Tian, Y.N. Procyanidins B2 reverses the T-2 toxin-induced mitochondrial apoptosis in TM3 leydig cells. J. Funct. Foods 2018, 45, 118–128. [Google Scholar] [CrossRef]
- Yang, L.; Tu, D.; Wang, N.; Deng, Z.; Zhan, Y.; Liu, W.; Hu, Y.; Liu, T.; Tan, L.; Li, Y.; et al. The protective effects of DL-Selenomethionine against T-2/HT-2 toxins-induced cytotoxicity and oxidative stress in broiler hepatocytes. Toxicol. In Vitro 2019, 54, 137–146. [Google Scholar] [CrossRef]
- Chaudhary, M.; Rao, P.V. Brain oxidative stress after dermal and subcutaneous exposure of T-2 toxin in mice. Food Chem. Toxicol. 2010, 48, 3436–3442. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Zhai, N.; Chen, X.; Gan, F.; Li, H.; Huang, K. Ochratoxin A-induced apoptosis of IPEC-J2 cells through ROS-mediated mitochondrial permeability transition pore opening pathway. J. Agric. Food Chem. 2017, 65, 10630–10637. [Google Scholar] [CrossRef]
- Fatima, Z.; Guo, P.; Huang, D.; Lu, Q.; Wu, Q.; Dai, M.; Cheng, G.; Peng, D.; Tao, Y.; Ayub, M.; et al. The critical role of p16/Rb pathway in the inhibition of GH3 cell cycle induced by T-2 toxin. Toxicology 2018, 400–401, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.J.; Sanderson, L.E.; Crosier, K.E.; Crosier, P.S. Mitochondrial metabolism, reactive oxygen species, and macrophage function-fishing for insights. J. Mol. Med. 2014, 92, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.H.; Dasgupta, A.; Ding, J.; Indig, F.E.; Ghosh, P.; Longo, D.L. Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J. 2014, 28, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Sesaki, H.; Kageyama, Y.; Reddy, P.H. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim. Biophys. Acta 2012, 1822, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Gao, Y.; Zhang, C.; Li, H.; Pan, S.; Wang, X.; Du, S.; Deng, Z.; Wang, L.; Song, Z.; et al. SAMM50 Affects Mitochondrial Morphology through the Association of Drp1 in Mammalian Cells. FEBS Lett. 2016, 590, 1313–1323. [Google Scholar] [CrossRef]
- Böhler, P.; Stuhldreier, F.; Anand, R.; Kondadi, A.K.; Schlütermann, D.; Berleth, N.; Deitersen, J.; Wallot-Hieke, N.; Wu, W.; Frank, M.; et al. The mycotoxin phomoxanthone A disturbs the form and function of the inner mitochondrial membrane. Cell Death Dis. 2018, 9, 286. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.H.; Thompson, C.B. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000, 7, 1182–1191. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Velkov, T.; Tang, S.; Dai, C. T-2 toxin-induced toxicity in neuroblastoma-2a cells involves the generation of reactive oxygen, mitochondrial dysfunction and inhibition of Nrf2/HO-1 pathway. Food Chem. Toxicol. 2018, 114, 88–97. [Google Scholar] [CrossRef]
- Yang, L.; Tu, D.; Zhao, Z.; Cui, J. Cytotoxicity and apoptosis induced by mixed mycotoxins (T-2 and HT-2 toxin) on primary hepatocytes of broilers in vitro. Toxicon 2017, 129, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Zunino, R.; Gangaraju, S.; Rippstein, P.; McBride, H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J. Biol. Chem. 2005, 280, 25060–25070. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B.; et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, M.; Alam, M.M.; Tsushima, A.; Yoshizawa, T.; Sakato, N. Apoptosis induction by T-2 toxin: Activation of caspase-9, caspase-3, and DFF-40/CAD through cytosolic release of cytochrome c in HL-60 cells. Biosci. Biotechnol. Biochem. 2001, 65, 1741–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renault, T.T.; Floros, K.V.; Elkholi, R.; Corrigan, K.A.; Kushnareva, Y.; Wieder, S.Y.; Lindtner, C.; Serasinghe, M.N.; Asciolla, J.J.; Buettner, C.; et al. Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol. Cell 2015, 57, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title | GenBank Accession Number | 5’–3’ Primer Sequence | PCR Product/bp |
|---|---|---|---|
| Caspase 3 | NM_004346.3 | F: atgctgaaacagtatgccgacaa | 98 |
| R: gcgtcaaaggaaaaggactcaaat | |||
| Caspase 7 | BC015799.1 | F: tgacttcctcttcgcctattccac | 124 |
| R: gatttccaggtcttttccgtgc | |||
| Caspase 9 | NM_001229.4 | F: agccaaccctagaaaaccttacc | 115 |
| R: tcaccaaatcctccagaaccaat | |||
| Bax | NM_004324.3 | F: ccccgagaggtctttttccgag | 113 |
| R: agggccttgagcaccagtttg | |||
| Bcl-2 | BC027258.1 | F: cctctgtttgatttctcctggc | 153 |
| R: ttctactgctttagtgaaccttttg | |||
| cytochrome c | NM_018947.5 | F: cttcagaaataaggaaatagggga | 154 |
| R: caaaataagcatgtaggtggca | |||
| Fis1 | NM_016068.2 | F: gcacgcagtttgagtacgcct | 113 |
| R: ctgttcctccttgctccctttg | |||
| Mfn1 | NM_033540.2 | F: ttgagagatgacctggtgttagtag | 130 |
| R: ttagtgttgattcagagtttgcgac | |||
| Mfn2 | BC017061.1 | F: tctttatgctgatgttgagttttgg | 159 |
| R: tttgggagaggtgttgcttattt | |||
| Drp1 | BC024590.1 | F: gcatcacatcagagattgtttacca | 113 |
| R: tagcacttttatcatccacgggtt | |||
| OPA1 | BC075805.1 | F: aatgactttgcggaggacagc | 145 |
| R: ttgcattttcagtatccttgagacg | |||
| VDAC1 | NM_003374 | F: accgagattactgtggaaga | 75 |
| R: agtgttaggtgagaaggatga | |||
| β-actin | NM_001101.3 | F: gcatgggtcagaaggattcc | 276 |
| R: tggatagcaacgtacatggc |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Guo, W.; Wang, J.; Yang, X.; Zhang, Z.; Zhao, Z. T-2 Toxin-Induced Oxidative Stress Leads to Imbalance of Mitochondrial Fission and Fusion to Activate Cellular Apoptosis in the Human Liver 7702 Cell Line. Toxins 2020, 12, 43. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010043
Yang J, Guo W, Wang J, Yang X, Zhang Z, Zhao Z. T-2 Toxin-Induced Oxidative Stress Leads to Imbalance of Mitochondrial Fission and Fusion to Activate Cellular Apoptosis in the Human Liver 7702 Cell Line. Toxins. 2020; 12(1):43. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010043
Chicago/Turabian StyleYang, Junhua, Wenbo Guo, Jianhua Wang, Xianli Yang, Zhiqi Zhang, and Zhihui Zhao. 2020. "T-2 Toxin-Induced Oxidative Stress Leads to Imbalance of Mitochondrial Fission and Fusion to Activate Cellular Apoptosis in the Human Liver 7702 Cell Line" Toxins 12, no. 1: 43. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010043