Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate



Laboratory of Pathophysiology, Department of Biomedical Sciences, University of Antwerp, 2000 Antwerpen, Belgium
*
Author to whom correspondence should be addressed.
Toxins 2020, 12(1), 58; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010058
Submission received: 18 December 2019
/
Revised: 13 January 2020
/
Accepted: 17 January 2020
/
Published: 19 January 2020
(This article belongs to the Special Issue Comorbidities in Chronic Kidney Disease (CKD))
Abstract
:The protein-bound uremic toxins, indoxyl sulfate (IS) and p-cresyl sulfate (PCS), are considered to be harmful vascular toxins. Arterial media calcification, or the deposition of calcium phosphate crystals in the arteries, contributes significantly to cardiovascular complications, including left ventricular hypertrophy, hypertension, and impaired coronary perfusion in the elderly and patients with chronic kidney disease (CKD) and diabetes. Recently, we reported that both IS and PCS trigger moderate to severe calcification in the aorta and peripheral vessels of CKD rats. This review describes the molecular and cellular mechanisms by which these uremic toxins induce arterial media calcification. A complex interplay between inflammation, coagulation, and lipid metabolism pathways, influenced by epigenetic factors, is crucial in IS/PCS-induced arterial media calcification. High levels of glucose are linked to these events, suggesting that a good balance between glucose and lipid levels might be important. On the cellular level, effects on endothelial cells, which act as the primary sensors of circulating pathological triggers, might be as important as those on vascular smooth muscle cells. Endothelial dysfunction, provoked by IS and PCS triggered oxidative stress, may be considered a key event in the onset and development of arterial media calcification. In this review a number of important outstanding questions such as the role of miRNA’s, phenotypic switching of both endothelial and vascular smooth muscle cells and new types of programmed cell death in arterial media calcification related to protein-bound uremic toxins are put forward and discussed.
Keywords:
uremic toxins; arterial calcification; lipid metabolism; inflammation; coagulation; endothelial dysfunction; epigeneticsKey Contribution: Indoxyl sulfate and p-cresyl sulfate promote arterial calcification, and this is linked to a complex interplay between inflammation, coagulation, and lipid metabolism pathways.
1. Introduction
During chronic kidney disease (CKD), uremic retention solutes accumulate in the bloodstream due to progressive kidney function loss. Three classes of uremic retention solutes exist: (i) low-molecular-weight water-soluble solutes (<500 Da), (ii) middle-molecular-weight solutes (>500 Da), and (iii) protein-bound solutes. This latter class is characterized by a limited dialytic removal due to the high molecular weight of the protein complexes that complicates their movement across the dialysis membrane [1]. Both indoxyl sulfate (IS) and p-cresyl sulfate (PCS) belong to the protein-bound uremic toxins and originate from protein fermentation in the intestine. The intestinal microbiota facilitates the breakdown of tyrosine/phenylalanine and tryptophan into, respectively, p-cresol and indole, which are absorbed and detoxified by oxidation and conjugation with sulfate [2]. In the bloodstream, IS and PCS bind to albumin, which implies that glomerular filtration does not take place and thus requires tubular transporter systems in the kidney to excrete these two protein-bound uremic toxins. Basolateral organic anion transporter 1 (OAT1) and 3 (OAT3), breast cancer resistance protein (BCRP) and multidrug resistance protein 4 (MRP4) belong to the IS and PCS tubular transport system [3]. Due to progressive kidney function loss, the concentration of IS and PCS increases with CKD stage in humans, as shown in Table 1 (adapted from Lin et al., J. Food Drug Anal., 2019 [4]), ending up with levels of around 20-fold CKD stage 1. Moreover, free IS and PCS levels are 100-fold higher in pretreatment hemodialysis patients as compared to normal subjects [5]. There is, however, a high inter-individual variability, reflected by high standard deviations, which is also observed in other studies [6,7,8]. An explanation might be (i) alterations in colon microbiome attributed in part to dietary restrictions in CKD patients [9], (ii) modulation of IS and PCS transporters [10] and (iii) residual kidney function [11]. This inter-patient variability might influence the interpretation on the association between of IS and PCS concentrations and clinical outcomes. However, unbound PCS serum levels are suggested to hold a substantial predictive value for the survival in CKD patients [8]. Moreover, IS and PCS serum levels are also associated with cardiovascular disease and mortality [12,13]. Furthermore, Shafi et al. reported no association between total IS and PCS serum concentrations with cardiac death, sudden cardiac death, and first cardiovascular event [14]. These conflicting clinical associations demand for experimental study designs to unravel the role of IS and PCS in cardiovascular disease, which is crucial, as cardiovascular defects account for 50% of all deaths in CKD patients [15], having high serum IS and PCS levels.
2. Molecular Mechanisms by Which IS and PCS Induce Vascular Calcification
Arterial media calcification is a life-threatening disease that manifests in elderly and patients with chronic kidney disease (CKD) and diabetes mellitus. The disease phenotype is characterized by a passive and active deposition of calcium phosphate crystals in the media layer of the arterial wall that leads to arterial stiffness, which in turn induces hypertension, left ventricular hypertrophy, and impaired coronary perfusion. Arterial media calcification occurs already in the early stages of CKD, and more than half of the CKD patients on dialysis suffer from it [16,17]. Moreover, calcifications in the arterial wall are also present in children with CKD [18]. Moreover, CKD patients who also suffer from diabetes mellitus have a higher incidence of arterial calcification compared to nondiabetic hemodialysis patients [19]. Poor control of glucose levels is a predictor of arterial calcification in humans [19,20]. Our laboratory recently reported that both IS and PCS are important harmful vascular toxins, as they trigger moderate to severe arterial media calcification in CKD rats, which goes along with the activation of inflammation (i.e., acute phase response signaling pathway) and coagulation (i.e., intrinsic/extrinsic prothrombin activation pathway) pathways linked with increased circulating glucose levels and insulin resistance. These changes were even observed after four days of IS or PCS exposure, i.e., before arterial media calcifications had developed, indicating that the IS/PCS mediated upregulation of inflammation and coagulation precedes the vascular calcification process [21]. Additionally, in this study, escape from uremic-toxin-induced calcification was linked with liver X receptor and farnesoid X/liver X receptor signaling pathways, discussed more in detail below. This review focuses on these signaling pathways, as well as on the connection with endothelial dysfunction and the effects on microRNAs.
2.1. Inflammation and Coagulation Signaling Pathways
The inflammatory acute phase response signaling pathway is a physiological host-defense mechanism to injury, including trauma, acute infection, and myocardial infarction [22]. In addition, a low-grade chronic acute phase response exists and is characterized by chronically elevated levels of acute phase proteins in response to metabolically triggered inflammation [23]. During CKD, the human body undergoes a permanent status of low-grade inflammation. The uremic retention solutes IS and PCS regulate inflammation in multiple cell types, including adipocytes, endothelial cells, macrophages, proximal tubular cells, and glial cells [24,25,26,27,28]. Inflammatory responses are associated with the development of arterial calcification [29]. Moreover, a recent study in 112 chronic hemodialysis patients correlated acute-phase proteins (i.e., C-reactive protein, ferritin, hepcidin, and albumin) with the development of abdominal aortic calcification [30]. In addition, the acute-phase proteins serum amyloid A and C-reactive protein have been reported not to be solely produced by hepatocytes, but also in the arterial wall, by vascular smooth muscle cells (VSMCs). Both proteins stimulate the phenotypic switch of VSMCs into bone-like cells through activation of the p38 MAPK pathway and oxidative stress pathways [31,32]. As mentioned above, our study also revealed that, after proteomic analysis of aortic tissue from either IS or PCS exposed CKD rats, coagulation pathways (intrinsic/extrinsic prothrombin activation pathways) play a central role in the arterial calcification process [21]. A close link between coagulation and inflammation exists as coagulation factors, such as fibrinogens and prothrombin, also belong to the acute phase proteins [22]. A recent study showed that IS induces platelet hyperactivity, with an elevated response to collagen and thrombin, through activation of the p38 MAPK and oxidative stress pathways [33]. Moreover, other studies reported an association between increased serum IS levels and thrombotic complications in CKD patients [33,34]. Then again, the thrombin–antithrombin complex level, used as a marker for thrombin formation in vivo, correlates with the presence and severity of coronary artery calcification [35,36]. An in vitro study of Kapustin et al. showed that Gla-containing coagulation factors (i.e., prothrombin and protein C and S) inhibit the vascular calcification process [37]. This might mean that the calcification-inducing effects of warfarin [38,39] can also be ascribed to its anti-coagulant actions, next to the fact that it prevents the production of the active form of matrix Gla protein, an important endogenous calcification inhibitor (warfarin inhibits carboxylation of Gla-proteins by inhibiting vitamin K recycling). All of these data suggest that coagulation and vascular calcification are interconnected and that the uremic toxins IS and PCS could play an important role herein. However, a recent multicenter randomized controlled trial showed that withdrawal of vitamin K antagonists in hemodialysis patients did not influence progression of arterial calcification progression after 18 months [40]. These conflicting data again indicate that the arterial calcification process is the result of a complex interplay between different pathological pathways.
Interestingly, in our experimental study, a minority (3 out of 15) of CKD rats exposed to either IS or PCS did not develop arterial media calcification. This escape from uremic toxin-induced arterial calcification was related to liver X receptor and farnesoid X/liver X receptor signaling pathways, an important pathway in lipid metabolism. Activation of the liver X receptor (LXR) by the agonist GW3965 leads to anti-inflammatory actions in endothelial cells and macrophages by attenuation of the NF-kB pathway and IL-8 production [41,42]. Moreover, LXR activation inhibits cardiomyocyte apoptosis by restored mitochondrial membrane potential level and thus decreased reactive oxygen species (ROS) production [43]. These studies suggest that IS- and PCS-induced vascular calcification could be counteracted by preventing inflammation and oxidative stress events through the LXR pathway. This is further strengthened by results from our study in which, next to upregulation of acute phase response (inflammation), also oxidative stress pathways, i.e., Glutathione Mediated Detoxification and Glutathione Redox Reactions I, were observed in calcified aortic tissue of rats exposed to either IS or PCS for 7 weeks [21]. Furthermore, epigenetic involvement, often influenced by inflammation and oxidative stress, adds to the complexity by which uremic toxins trigger cardiovascular complications in CKD patients. IS has been associated with the regulating mammalian methyltransferase Set7/9, an epigenetic inducer of inflammatory genes, in VSMCs [44]. Moreover, data from an experimental rat study showed that IS exposure induces arterial thrombosis via decreased aortic levels of sirtuin 1, a class III histone deacetylase involved in oxidative stress [45]. We conducted a curated chemical and genomic/proteomic perturbagen matching analysis to predict upstream regulators that could be responsible for the observed changes in the arterial proteins linked to inflammation and coagulation signaling pathways. This analysis revealed a major role for altered energy and glucose metabolism, including elevated glucose levels and insulin receptor dysfunction [21]. This is interesting in view of the fact that diabetes increases the risk for vascular calcification in CKD patients [19]. Koppe et al. showed that chronic PCS exposure promoted insulin resistance and hyperglycemia in CKD mice [46]. Taken together, our study [21] suggests that IS and PCS stimulate the aortic media calcification via alterations in glucose metabolism, which in turn may stimulate inflammation, coagulation, and oxidative stress pathways in the aorta. It is worth noting that other studies have reported that the aryl hydrocarbon receptor, activated by IS, also regulates coagulation in VSMCs [47] and endothelial cells [48]. Interestingly, transient hyperglycemia also triggers Set7/9-mediated epigenetic changes in the promoter of NF-kB subunit p65, favoring overexpression of inflammatory genes [49]. Moreover, hyperglycemia favors the downregulation of sirtuin 1 in endothelial cells, and, by this, it upregulates vascular p66She gene transcription, which is implicated in mitochondrial ROS production and induction of apoptotic cell death. Moreover, metformin, an antidiabetic drug, has been reported to stimulate sirtuin 1 activation, which suppressed the increase of poly (ADP-ribose) polymerase (PARP) mediated mitochondrial ROS and thereby halted oxidative stress and inflammation in the retina of diabetic rats [50]. Remarkably, both metformin [51] and minocycline (PARP-inhibitor) [52] inhibit the development of arterial calcification in rats, pointing to the importance of oxidative stress and cell death events in the process of vascular calcification. Figure 1 gives a schematic overview of the interplay between inflammation, coagulation, and lipid metabolism and the role of epigenetics in IS and PCS-induced arterial calcification.
2.2. MicroRNAs, Upcoming Important Epigenetic Regulators in Uremic Toxins-Induced Vascular Calcification
MicroRNAs, small noncoding RNAs, are approximately 18–25 nucleotides long and regulate the protein expression of the target mRNA without affecting the gene sequence [53]. It has been well established that microRNAs play an important role in the vascular calcification process [54]. Protein-bound uremic toxins stimulate the expression of microRNA miR-92a in endothelial cells and, by this, suppress the gene expression of sirtuin 1, Krüppel-like factors 2 and 4, and endothelial nitric oxide synthase (eNOS) [55]. Interestingly, nitric oxide (NO) may be protective against arterial calcification, as in vitro NO inhibits murine VSMCs calcification and osteochondrogenic transdifferentiation via inhibition of TGFβ-induced phosphorylation of SMAD2/3 [56] and metformin halts arterial calcification through restoration of NO bioavailability (via the AMPK-eNOS-NO pathway) in rats [51,57]. As already mentioned above, metformin also increases the expression of sirtuin 1 [50] and thus might be able to counteract the IS/PCS-miR-92a triggered suppression of sirtuin 1 and, by this, decrease oxidative stress and inflammation events.
Another microRNA, miR-29b, has been reported to influence the development of arterial calcification in VSMC cell cultures and 5/6th nephrectomized rats [58]. Moreover, a recent study showed that miR-29b was downregulated in human aortic smooth muscle cells in which IS induced calcification, and by this increased Wnt7b/β catenin signaling [59]. IS also promotes renal fibrosis by DNA hypermethylation of sFRP5, leading to activation of the Wnt/β catenin signaling pathway [60]. Interestingly, our laboratory found that sclerostin, a Wnt/β catenin inhibitor, might be linked to prevention of vascular calcification development [39]. Furthermore, uremic toxins also correlate with elevated serum levels of miR-126, miR-143, miR-145, and miR-223 [61]. This latter microRNA, when overexpressed, triggers the uptake of glucose via the glucose transporter GLUT4 in cardiomyocytes [62] and stimulates the arterial calcification process by acting on the VSMC phenotypic switch [63], which is further discussed in the next paragraph. For this reason, miR-223 might have played a role in the hyperglycemia seen in our previous study concomitantly with the development of IS/PCS induced aortic media calcification. This, in turn, could have stimulated inflammation, coagulation, and oxidative stress pathways in the aorta. Again, this points to an important role of hyperglycemia in the toxic effects of IS and PCS on the vasculature.
2.3. Novel Research Fields to be Explored
2.3.1. Protein-Bound Uremic Toxins Influence Phenotypic Switch of Multiple Cell Types
The phenotypic transition of VSMCs into osteo/chondrogenic cells is a hallmark of the arterial calcification process [64]. Studies have shown that both IS and PCS are able to induce osteo/chondrogenic transdifferentiation of VSMCs by downregulation of smooth muscle genes (i.e., smooth muscle 22α, α-smooth muscle actin) and upregulation of bone-like genes (i.e., runx2, alkaline phosphatase, and osteopontin) [65,66]. This genotypic switching stimulates the VSMCs to produce calcifying exosomes, in which calcium phosphate crystals aggregate and, by this, mineralize the extracellular matrix [64]. Interestingly, IS and PCS also induce the transdifferentiation of other cell types, such as proximal renal tubular cells. When these cells are exposed to either IS or PCS, an epithelial to mesenchymal transition (EMT) occurs. During EMT, proximal renal tubular cells lose their adhesive characteristics with downregulation of E-cadherin in favor of mesenchymal fibroblast-like characteristics with upregulation of α-smooth muscle actin, fibronectin, N-cadherin, and vimentin. Several studies have shown that exposure to IS or PCS induces these phenotypic alterations, leading to glomerular sclerosis and interstitial fibrosis, with further stimulation of the progression of CKD [67,68,69]. Moreover, AST120, an oral spherical carbonaceous adsorbent, absorbs the precursors of IS and PCS and ameliorates the EMT process in renal tubular cells, and this is correlated with a decrease of serum IS levels [70]. Both TGFβ/Smad signaling and β-catenin signaling have been reported to act as main regulators of the uremic-toxin-induced EMT process in renal tubular cells [69,71]. In addition to EMT, endothelial to mesenchymal transition (EndMT), a subtype of EMT, involves the transdifferentiation of endothelial cells into mesenchymal stem-like cells which can differentiate further into multiple cell lineages: fibroblasts/myofibroblasts, osteoblasts/osteocytes, chondrocytes, and/or adipocytes [72]. Various studies have linked EndMT to the development of arterial calcification [73,74,75]. Its specific role in the calcification process, however, is not yet completely understood. It would be interesting to investigate whether IS and PCS are involved in the EndMT process, as (i) IS and PCS are known to promote arterial media calcification, and (ii) IS and PCS stimulate the EMT process in proximal tubular cells. Moreover, research so far predominantly focused on the transdifferentiation of VSMCs into bone-like cells, whilst the phenotypic transition of endothelial cells (EndMT) is often neglected and might be more important than generally thought [76]. Subsequently, in vitro experiments have demonstrated that both IS and PCS induce endothelial dysfunction, underlining even more that the endothelium plays a crucial role during uremic-toxin-induced arterial media calcification.
2.3.2. The Endothelium, an Overlooked Structure in the Process of Uremic Toxin Induced Vascular Calcification
IS and PCS induce deleterious effects in the endothelium, including the inhibition of cell proliferation and wound healing, and the increase in oxidative stress responses, cell senescence, and the release of endothelial microparticles [77,78,79,80]. A prospective observational study in 41 CKD patients revealed that AST-120 treatment decreased serum IS levels, as well as the oxidized/reduced glutathione ratio [79]. Interestingly, a depletion of glutathione triggers ferroptotic events [81], a novel type of iron-mediated programmed cell death characterized by the accumulation of lipid peroxides. Moreover, IS-generated oxidative stress in human umbilical vein endothelial cells induces endothelial senescence [79,82]. Additionally, in other cell types, including proximal renal tubular cells and VSMCs, IS has been reported to accelerate the process of cell senescence, as evidenced by the upregulation of p53 activity [83,84]. Activation of this tumor-suppressor protein p53 in the cell initiates apoptosis, senescence, and ferroptosis [85]. Which of these cell fates eventually occurs depends on yet unknown mechanisms that direct allow p53 to selectively activate downstream targets. The involvement of cell senescence and apoptosis in arterial calcification has been reported [86,87]. To which extent ferroptosis is involved in the calcification process remains unknown and, therefore, could be a new research field that is worth being explored, in order to further unravel the mechanisms underlying the effects of IS and PCS, as well as other toxins on the development of vascular calcification.
An additional challenging approach would consist in a further in-depth investigation at which extent the IS-induced release of microparticles derived from the endothelial cell membranes plays a role in the development of arterial calcification [88]. These endothelial microparticles (EMPs) have pro-inflammatory and pro-coagulant characteristics by interacting with monocytes. Moreover, EMPs accumulate different substances which influence the behavior of recipient cells. Buendia et al. reported that endothelial dysfunction favors the release of EMPs with a high content of calcium and bone morphogenetic protein-2, two important stimulators of the osteo/chondrogenic transdifferentiation of VSMCs [89]. In addition, microRNAs can be released via EMPs and influence endothelial function, i.e., through modulation of eNOS bioavailability, as mentioned above. Figure 2 gives an overview of the IS- and PCS-induced effects on VSMCs and endothelial cells.
3. Concluding Remarks
In conclusion, the protein-bound uremic toxins IS and PCS are considered to be harmful vascular toxins. Their vascular toxicity is associated with the upregulation of inflammation, coagulation, and oxidative stress pathways. Moreover, hyperglycemia seems to be an important player in these events. On the other hand, escape from IS/PCS-induced arterial media calcification was linked to activation of lipid metabolism. This suggests that the balance in glucose versus lipid metabolism could be crucial in uremic-toxin-induced vascular calcification.
Author Contributions
Drafted and revised the manuscript, B.O.; Revised the manuscript, P.C.D. and A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fund of Scientific Research-Flanders (FWO) grant number 1S22217N. The APC was funded by Laboratory of Pathophysiology.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, H.A.; van den Heuvel, L.P.; Ringens, L.H.; Dankers, A.C.; Russel, F.G.; Wetzels, J.F.; Hoenderop, J.G.; Masereeuw, R. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS ONE 2011, 6, e18438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.N.; Wu, I.W.; Huang, Y.F.; Peng, S.Y.; Huang, Y.C.; Ning, H.C. Measuring serum total and free indoxyl sulfate and p-cresyl sulfate in chronic kidney disease using UPLC-MS/MS. J. Food Drug Anal. 2019, 27, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirich, T.L.; Funk, B.A.; Plummer, N.S.; Hostetter, T.H.; Meyer, T.W. Prominent accumulation in hemodialysis patients of solutes normally cleared by tubular secretion. J. Am. Soc. Nephrol. 2014, 25, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephro. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Eloot, S.; Van Biesen, W.; Roels, S.; Delrue, W.; Schepers, E.; Dhondt, A.; Vanholder, R.; Glorieux, G. Spontaneous variability of pre-dialysis concentrations of uremic toxins over time in stable hemodialysis patients. PLoS ONE 2017, 12, e0186010. [Google Scholar] [CrossRef]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transpl. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B.; et al. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef]
- Hsueh, C.H.; Yoshida, K.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.M.; Giacomini, K.M. Identification and Quantitative Assessment of Uremic Solutes as Inhibitors of Renal Organic Anion Transporters, OAT1 and OAT3. Mol. Pharm. 2016, 13, 3130–3140. [Google Scholar] [CrossRef]
- Toth-Manikowski, S.M.; Sirich, T.L.; Meyer, T.W.; Hostetter, T.H.; Hwang, S.; Plummer, N.S.; Hai, X.; Coresh, J.; Powe, N.R.; Shafi, T.; et al. Contribution of ‘clinically negligible’ residual kidney function to clearance of uremic solutes. Nephrol. Dial. Transpl. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Pawlak, D. Indoxyl sulfate - the uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Wu, V.; Wu, P.C.; Wu, C.J. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Sirich, T.L.; Meyer, T.W.; Hostetter, T.H.; Plummer, N.S.; Hwang, S.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R.; et al. Results of the HEMO Study suggest that p-cresol sulfate and indoxyl sulfate are not associated with cardiovascular outcomes. Kidney Int. 2017, 92, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transpl. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney. Dis. 2004, 44, 1024–1030. [Google Scholar] [CrossRef]
- Sigrist, M.K.; Taal, M.W.; Bungay, P.; McIntyre, C.W. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 1241–1248. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.C.; Mitsnefes, M.M. Cardiovascular disease in CKD in children: update on risk factors, risk assessment, and management. Am. J. Kidney. Dis. 2009, 54, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Ishimura, E.; Okuno, S.; Kitatani, K.; Kim, M.; Shoji, T.; Nakatani, T.; Inaba, M.; Nishizawa, Y. Different risk factors for peripheral vascular calcification between diabetic and non-diabetic haemodialysis patients--importance of glycaemic control. Diabetologia 2002, 45, 1446–1448. [Google Scholar] [CrossRef] [Green Version]
- Katz, R.; Budoff, M.J.; O’Brien, K.D.; Wong, N.D.; Nasir, K. The metabolic syndrome and diabetes mellitus as predictors of thoracic aortic calcification as detected by non-contrast computed tomography in the Multi-Ethnic Study of Atherosclerosis. Diabetic Med. 2016, 33, 912–919. [Google Scholar] [CrossRef] [Green Version]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Mare, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E.; et al. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Gruys, E.; Toussaint, M.J.; Niewold, T.A.; Koopmans, S.J. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. Sci. B 2005, 6, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Venteclef, N.; Jakobsson, T.; Steffensen, K.R.; Treuter, E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrin. Met. 2011, 22, 333–343. [Google Scholar] [CrossRef]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Saldanha, J.F.; Yi, D.; Mafra, D.; Fouque, D.; Soulage, C.O. The uremic toxin indoxyl sulfate exacerbates reactive oxygen species production and inflammation in 3T3-L1 adipose cells. Free Radical Res. 2016, 50, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Bessueille, L.; Magne, D. Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell. Mol. Life Sci. 2015, 72, 2475–2489. [Google Scholar] [CrossRef]
- Avramovski, P.; Avramovska, M.; Sotiroski, K.; Sikole, A. Acute-phase proteins as promoters of abdominal aortic calcification in chronic dialysis patients. Saudi J. Kidney Dis. Transpl. 2019, 30, 376–386. [Google Scholar] [CrossRef]
- Henze, L.A.; Luong, T.T.D.; Boehme, B.; Masyout, J.; Schneider, M.P.; Brachs, S.; Lang, F.; Pieske, B.; Pasch, A.; Eckardt, K.U.; et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging 2019, 11, 5445–5462. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Wang, S. Serum Amyloid a Induces a Vascular Smooth Muscle Cell Phenotype Switch through the p38 MAPK Signaling Pathway. Biomed Res. Int. 2017, 2017, 4941379. [Google Scholar] [CrossRef]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Hsieh, M.Y.; Hung, S.C.; Kuo, K.L.; Tsai, T.H.; Lai, C.L.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C.; et al. Serum Indoxyl Sulfate Associates with Postangioplasty Thrombosis of Dialysis Grafts. J. Am. Soc. Nephrol. 2016, 27, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Spronk, H.M.; ten Cate, H.; Hofstra, L. Accelerated in vivo thrombin formation independently predicts the presence and severity of CT angiographic coronary atherosclerosis. JACC Cardiovasc. Imag. 2012, 5, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Horn, P.; Erkilet, G.; Veulemans, V.; Kropil, P.; Schurgers, L.; Zeus, T.; Heiss, C.; Kelm, M.; Westenfeld, R. Microparticle-Induced Coagulation Relates to Coronary Artery Atherosclerosis in Severe Aortic Valve Stenosis. PLoS ONE 2016, 11, e0151499. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Schoppet, M.; Schurgers, L.J.; Reynolds, J.L.; McNair, R.; Heiss, A.; Jahnen-Dechent, W.; Hackeng, T.M.; Schlieper, G.; Harrison, P.; et al. Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arter. Thromb. Vasc. Biol. 2017, 37, e22–e32. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, J.; Dhondt, A. The role of vitamin K in vascular calcification of patients with chronic kidney disease. Acta Clin. Belg. 2016, 71, 462–467. [Google Scholar] [CrossRef]
- De Mare, A.; Maudsley, S.; Azmi, A.; Hendrickx, J.O.; Opdebeeck, B.; Neven, E.; D’Haese, P.C.; Verhulst, A. Sclerostin as Regulatory Molecule in Vascular Media Calcification and the Bone-Vascular Axis. Toxins 2019, 11, 428. [Google Scholar] [CrossRef] [Green Version]
- De Vriese, A.S.; Caluwe, R.; Pyfferoen, L.; De Bacquer, D.; De Boeck, K.; Delanote, J.; De Surgeloose, D.; Van Hoenacker, P.; Van Vlem, B.; Verbeke, F.; et al. Multicenter Randomized Controlled Trial of Vitamin K Antagonist Replacement by Rivaroxaban with or without Vitamin K2 in Hemodialysis Patients with Atrial Fibrillation: the Valkyrie Study. J. Am. Soc. Nephrol. 2020, 31, 186–196. [Google Scholar] [CrossRef]
- Bi, X.; Song, J.; Gao, J.; Zhao, J.; Wang, M.; Scipione, C.A.; Koschinsky, M.L.; Wang, Z.V.; Xu, S.; Fu, G.; et al. Activation of liver X receptor attenuates lysophosphatidylcholine-induced IL-8 expression in endothelial cells via the NF-kappaB pathway and SUMOylation. J. Cell. Mol. Med. 2016, 20, 2249–2258. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Even-Or, O.; Xu, X.; van Rosmalen, M.; Lim, L.; Gadde, S.; Farokhzad, O.C.; Fisher, E.A. Nanoparticles containing a liver X receptor agonist inhibit inflammation and atherosclerosis. Adv. Healthc. Mater. 2015, 4, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhang, D.; Zhu, M.; Wang, Y.; Guo, S.; Xu, B.; Hou, G.; Feng, Y.; Liu, G. Liver X receptor alpha is targeted by microRNA-1 to inhibit cardiomyocyte apoptosis through a ROS-mediated mitochondrial pathway. Biochem. Cell. Biol. 2018, 96, 11–18. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.; Zhang, H.; Ning, Y.; Song, N.; Hu, J.; Cai, J.; Shi, Y.; Ding, X.; Zhang, X.; et al. Amelioration of Uremic Toxin Indoxyl Sulfate-Induced Osteoblastic Calcification by SET Domain Containing Lysine Methyltransferase 7/9 Protein. Nephron 2019, 141, 287–294. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D.; et al. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef] [Green Version]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallee, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [Green Version]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X.; et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012, 61, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xiao, J.; Li, R.; Qin, X.; Wang, F.; Mao, Y.; Liang, W.; Sheng, X.; Guo, M.; Song, Y.; et al. Metformin alleviates vascular calcification induced by vitamin D3 plus nicotine in rats via the AMPK pathway. Vasc. Pharmacol. 2016, 81, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138.e3113. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol.-Renal. 2014, 307, F891–F900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [Green Version]
- Kanno, Y.; Into, T.; Lowenstein, C.J.; Matsushita, K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc. Res. 2008, 77, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Sambe, T.; Mason, R.P.; Dawoud, H.; Bhatt, D.L.; Malinski, T. Metformin treatment decreases nitroxidative stress, restores nitric oxide bioavailability and endothelial function beyond glucose control. Biomed. Pharmacother. 2018, 98, 149–156. [Google Scholar] [CrossRef]
- Du, Y.; Gao, C.; Liu, Z.; Wang, L.; Liu, B.; He, F.; Zhang, T.; Wang, Y.; Wang, X.; Xu, M.; et al. Upregulation of a disintegrin and metalloproteinase with thrombospondin motifs-7 by miR-29 repression mediates vascular smooth muscle calcification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2580–2588. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Yu, Y.; Guan, X.; Nie, L.; Liu, Y.; He, T.; Xiong, J.; Xu, X.; Li, Y.; Yang, K.; Wang, Y.; et al. DNA hypermethylation of sFRP5 contributes to indoxyl sulfate-induced renal fibrosis. J. Mol. Med. 2017, 95, 601–613. [Google Scholar] [CrossRef]
- Massy, Z.A.; Metzinger-Le Meuth, V.; Metzinger, L. MicroRNAs Are Associated with Uremic Toxicity, Cardiovascular Calcification, and Disease. Contrib. Nephrol. 2017, 189, 160–168. [Google Scholar] [CrossRef]
- Lu, H.; Buchan, R.J.; Cook, S.A. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 2010, 86, 410–420. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; M’Baya-Moutoula, E.; Metzinger-Le Meuth, V.; Henaut, L.; Djelouat, M.S.; Benchitrit, J.; Massy, Z.A.; Metzinger, L. Inorganic phosphate accelerates the migration of vascular smooth muscle cells: evidence for the involvement of miR-223. PLoS ONE 2012, 7, e47807. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; De Schutter, T.M.; De Broe, M.E.; D’Haese, P.C. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011, 79, 1166–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transpl. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transpl. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Yu, M.A.; Ryu, E.S.; Jang, Y.H.; Kang, D.H. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab. Invest. 2012, 92, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [Green Version]
- Bolati, D.; Shimizu, H.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces epithelial-to-mesenchymal transition in rat kidneys and human proximal tubular cells. Am. J. Nephrol. 2011, 34, 318–323. [Google Scholar] [CrossRef]
- Bolati, D.; Shimizu, H.; Niwa, T. AST-120 ameliorates epithelial-to-mesenchymal transition and interstitial fibrosis in the kidneys of chronic kidney disease rats. J. Renal. Nutr. 2012, 22, 176–180. [Google Scholar] [CrossRef]
- Chang, L.C.; Sun, H.L.; Tsai, C.H.; Kuo, C.W.; Liu, K.L.; Lii, C.K.; Huang, C.S.; Li, C.C. 1,25(OH)2 D3 attenuates indoxyl sulfate-induced epithelial-to-mesenchymal cell transition via inactivation of PI3K/Akt/beta-catenin signaling in renal tubular epithelial cells. Nutrition 2019, 69, 110554. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, D.; Olsen, B.R. The role of endothelial-mesenchymal transition in heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Guo, Y.; Moon, J.H.; Jumabay, M.; Bostrom, K.I.; Yao, Y. Serine Protease Activation Essential for Endothelial-Mesenchymal Transition in Vascular Calcification. Circ. Res. 2015, 117, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Cubberly, M.R.; Bostrom, K.I. A role for the endothelium in vascular calcification. Circ. Res. 2013, 113, 495–504. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephro. 2011, 6, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Lu, L.; Hua, Y.; Huang, K.; Wang, I.; Huang, L.; Fu, Q.; Chen, A.; Chan, P.; Fan, H.; et al. Vasculopathy in the setting of cardiorenal syndrome: roles of protein-bound uremic toxins. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1–H13. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal. Bioanal. Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol.-Cell Physiol. 2012, 303, C126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. Senescence and dysfunction of proximal tubular cells are associated with activated p53 expression by indoxyl sulfate. Am. J. Physiol.-Cell Physiol. 2010, 299, C1110–C1117. [Google Scholar] [CrossRef] [PubMed]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Drozdov, I.; Shroff, R.; Beltran, L.E.; Shanahan, C.M. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ. Res. 2013, 112, e99–e109. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Buendia, P.; Montes de Oca, A.; Madueno, J.A.; Merino, A.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Rodriguez, M.; Carracedo, J. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. 2015, 29, 173–181. [Google Scholar] [CrossRef]
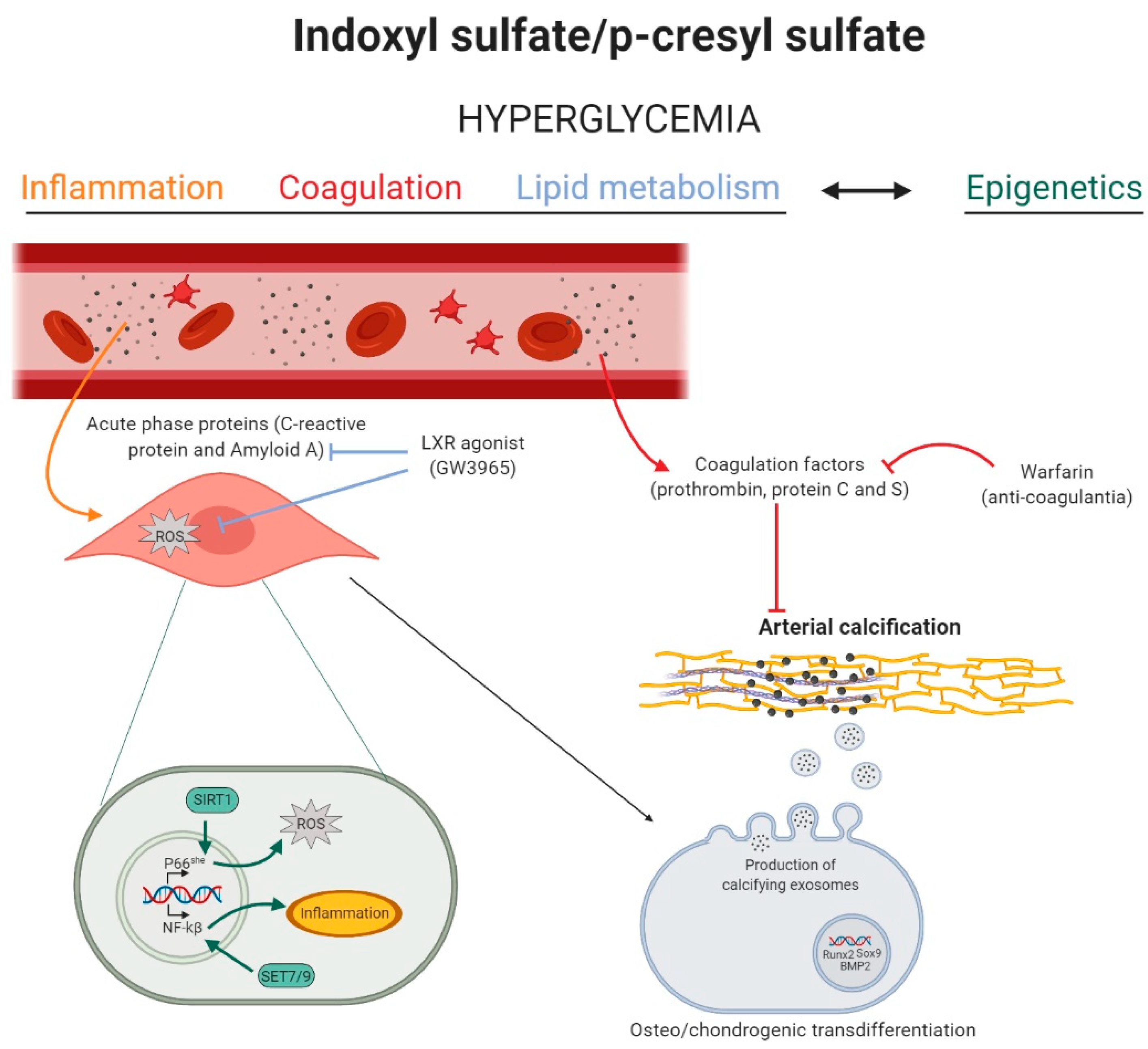
Figure 1.
The role of inflammation, coagulation, lipid metabolism, and epigenetics in indoxyl sulfate and p-cresyl sulfate induced arterial calcification. High levels of indoxyl sulfate (IS) and p-cresyl sulfate (PCS) induce a state of hyperglycemia, which activates inflammation, coagulation, lipid, and epigenetic pathways in the vascular cell. Inflammation (yellow): acute-phase response proteins induce reactive oxygen species (ROS) production in the vascular smooth muscle cell, stimulating the phenotypic switch into osteo-/chondrogenic cells. Coagulation (red): circulating coagulation factors inhibit arterial media calcification, while the anti-coagulant warfarin stimulates the calcification process. Lipid metabolism (blue): the liver-X-receptor (LXR) agonist blocks the inflammation mediated ROS production and, by this, inhibits arterial media calcification. Epigenetics (green): IS and PCS induced hyperglycemia might trigger sirtuin 1 (SIRT1)- and Set7/9-mediated epigenetic changes in the promoter of, respectively, p66She gene and NF-kB subunit p65, favoring, respectively, mitochondrial ROS production and inflammation in the cell. Figure was created with BioRender.com.
Figure 1.
The role of inflammation, coagulation, lipid metabolism, and epigenetics in indoxyl sulfate and p-cresyl sulfate induced arterial calcification. High levels of indoxyl sulfate (IS) and p-cresyl sulfate (PCS) induce a state of hyperglycemia, which activates inflammation, coagulation, lipid, and epigenetic pathways in the vascular cell. Inflammation (yellow): acute-phase response proteins induce reactive oxygen species (ROS) production in the vascular smooth muscle cell, stimulating the phenotypic switch into osteo-/chondrogenic cells. Coagulation (red): circulating coagulation factors inhibit arterial media calcification, while the anti-coagulant warfarin stimulates the calcification process. Lipid metabolism (blue): the liver-X-receptor (LXR) agonist blocks the inflammation mediated ROS production and, by this, inhibits arterial media calcification. Epigenetics (green): IS and PCS induced hyperglycemia might trigger sirtuin 1 (SIRT1)- and Set7/9-mediated epigenetic changes in the promoter of, respectively, p66She gene and NF-kB subunit p65, favoring, respectively, mitochondrial ROS production and inflammation in the cell. Figure was created with BioRender.com.

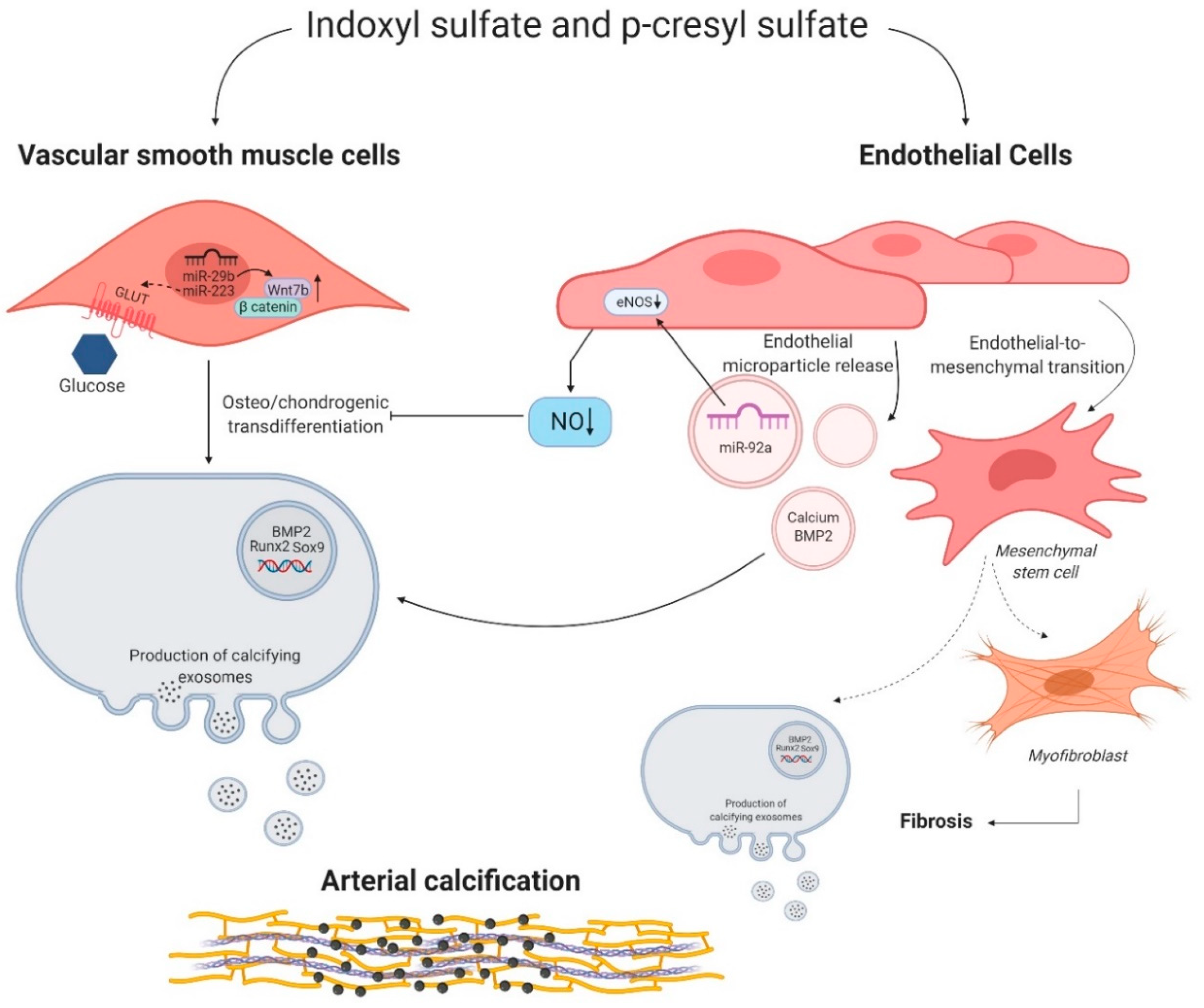
Figure 2.
Indoxyl sulfate and p-cresyl sulfate induced molecular mechanisms in vascular smooth muscle cells and endothelial cells. Indoxyl sulfate (IS) and p-cresyl sulfate (PCS) influence the behavior of vascular smooth muscle cells (VSMCs) and endothelial cells. Right side: microRNA miR-29b and miR-223 favor the osteo/chondrogenic switch of VSMCs by promoting the expression of Wnt7b/β catenin signaling and potentially increasing the uptake of glucose via a glucose transporter (GLUT), respectively. Left side: IS and PCS stimulate endothelial microparticle release. These microparticles secrete microRNA miR-92a, calcium, and bone morphogenic protein 2 (BMP2), which in turn induce a phenotypic switch of VSMCs into osteo/chondrogenic cells directly or indirectly through influencing endothelial nitric oxide synthase (eNOS) and thus decreasing nitric oxide (NO) bioavailability. Moreover, IS and PCS could trigger the endothelial to mesenchymal transition of endothelial cells into osteo/chondrogenic cells and myofibroblast, stimulating arterial calcification and fibrosis. Figure was created with BioRender.com.
Figure 2.
Indoxyl sulfate and p-cresyl sulfate induced molecular mechanisms in vascular smooth muscle cells and endothelial cells. Indoxyl sulfate (IS) and p-cresyl sulfate (PCS) influence the behavior of vascular smooth muscle cells (VSMCs) and endothelial cells. Right side: microRNA miR-29b and miR-223 favor the osteo/chondrogenic switch of VSMCs by promoting the expression of Wnt7b/β catenin signaling and potentially increasing the uptake of glucose via a glucose transporter (GLUT), respectively. Left side: IS and PCS stimulate endothelial microparticle release. These microparticles secrete microRNA miR-92a, calcium, and bone morphogenic protein 2 (BMP2), which in turn induce a phenotypic switch of VSMCs into osteo/chondrogenic cells directly or indirectly through influencing endothelial nitric oxide synthase (eNOS) and thus decreasing nitric oxide (NO) bioavailability. Moreover, IS and PCS could trigger the endothelial to mesenchymal transition of endothelial cells into osteo/chondrogenic cells and myofibroblast, stimulating arterial calcification and fibrosis. Figure was created with BioRender.com.


{kind=link}
{kind=link}
Table 1.
Serum levels of albumin bound (total) and unbound (free) IS and PCS.
| CKD | Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 |
|---|---|---|---|---|---|
| N | 29 | 49 | 64 | 40 | 22 |
| Total IS (mg/L) | 1.03 ± 0.85 | 1.54 ± 1.11 | 2.22 ± 1.79 | 4.74 ± 4.34 | 18.21 ± 15.06 |
| Total PCS (mg/L) | 2.69 ± 4.34 | 4.42 ± 4.47 | 6.45 ± 7.12 | 16.10 ± 13.98 | 27.00 ± 17.66 |
| Free IS (mg/L) | 0.08 ± 0.06 | 0.11 ± 0.09 | 0.17 ± 0.13 | 0.49 ± 0.72 | 2.36 ± 2.64 |
| Free PCS (mg/L) | 0.15 ± 0.20 | 0.24 ± 0.29 | 0.36 ± 0.37 | 1.36 ± 2.58 | 2.38 ± 2.03 |
Data represent the mean ± standard deviation.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010058
AMA Style
Opdebeeck B, D’Haese PC, Verhulst A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins. 2020; 12(1):58. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010058
Chicago/Turabian StyleOpdebeeck, Britt, Patrick C. D’Haese, and Anja Verhulst. 2020. "Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate" Toxins 12, no. 1: 58. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12010058
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.