Improved Sample Selection and Preparation Methods for Sampling Plans Used to Facilitate Rapid and Reliable Estimation of Aflatoxin in Chicken Feed

 , ,
, , 
Abstract
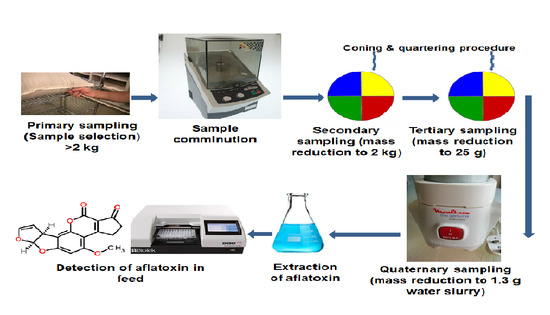
:
1. Introduction
2. Results and Discussion
2.1. Highlights of Major Modification of the Improved Aflatoxin Test Procedure
- (a)
- Primary sampling or sample selection (number and size of incremental samples, type of sampling tools for open and closed sub-lots, random procedure, size of incremental and aggregate samples, Section 4.1.1)
- (b)
- Secondary sampling (size of laboratory sample determined employing FAO Mycotoxin Sampling Tool, coning and quartering method improved by performing all coning and shoveling procedures under a steadfast funnel for mass-reduction of aggregate sample to 2 kg laboratory sample, Section 4.1.2)
- (c)
- Tertiary sampling (size of test portion determined employing FAO Mycotoxin Sampling Tool, the improved coning and quartering method in Point (b) for mass-reduction of laboratory sample to 25 g test portion, Section 4.1.3)
- (d)
- Quaternary sampling (homogenization and splitting of test portion by water slurring at matrix/water, 25:37.5, w/w; optimal matrix to organic solvent ratio for solid–liquid extraction, slurry/extraction solvent, 1.3:86.5, w/v, Section 4.1.4)
- (e)
- Quantification of AFB1 (optimal organic solvent to aqueous buffer ratio for AFB1 extraction back to aqueous phase-modified extract to aqueous buffer mixture was modified to 80% acetonitrile extraction solvent: PBS-T mixture, 100:650, v/v, Section 4.1.5)
2.2. Enzyme-Linked Immunosorbent Assay of Prepared Standards for Determination of Aflatoxin Content in Chicken Feed Samples
2.3. Validation Results of the Improved Aflatoxin Test Procedure
2.3.1. Method Accuracy and Precision
2.3.2. Limits of Detection (LOD) and Quantification (LOQ) Values
2.3.3. Linearity
2.3.4. Robustness and Ruggedness of the Aflatoxin Extraction Procedure
2.3.5. Evaluation of the Improved Aflatoxin Test Procedure
3. Conclusions
4. Materials and Methods
4.1. Description of Improved Aflatoxin Test Procedure
4.1.1. Sample Selection (Primary Sampling)
4.1.2. Comminution and Mass Reduction of Samples (Secondary Sampling)
4.1.3. Preparation of Test Portions (Tertiary Sampling)
4.1.4. Sample Extraction (Quaternary Sampling)
4.1.5. Quantification of AFB1 in Feed Samples
4.2. In-House Validation of Improved Aflatoxin Test Procedure
4.2.1. Materials
4.2.2. Study Design
4.2.3. Aflatoxin Spike and Recovery Studies
Comparison of Conventional (Dry) and Novel Water Slurry (Wet) Milling Procedure
Comparison of Different Extraction Conditions Associated with Wet Milling Procedure
4.2.4. Replication Studies
Estimation of Intra-Laboratory Variability (Repeatability)
Estimation of Intermediate Variability Associated with Wet Milling Procedure
4.2.5. Determination of Limits of Detection (LOD) and Quantification (LOQ)
4.2.6. Linearity Studies
4.2.7. Robustness and Ruggedness Studies
4.2.8. Evaluation of the Improved Aflatoxin Test Procedure
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oloo, R.G.; Okoth, S.; Wachira, P.; Mutiga, S.; Ochieng, P.; Kago, L.; Nganga, F.; Entfellner, J.B.D.; Ghimire, S. Genetic profiling of aspergillus isolates with varying aflatoxin production potential from different maize-growing regions of Kenya. Toxins 2019, 11, 467. [Google Scholar] [CrossRef] [Green Version]
- Benkerroum, N. Aflatoxins: Producing-molds, structure, health issues and incidence in Southeast Asian and Sub-Saharan African Countries. Int. J. Environ. Res. Public Health 2020, 17, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Mahato, D.K.; Kamle, M.; Mohanta, T.K.; Kang, S.G. Aflatoxins: A global concern for food safety, human health and their management. Front. Microbiol. 2017, 7, 2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.S.; Sara, C.C.; Fernandes, J.O. Prevalent mycotoxins in animal feed: Occurrence and analytical methods. Toxins 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Aflatoxins; REF. No.: WHO/NHM/FOS/RAM/18.1; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- World Health Organization; International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risks to humans. In Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene; World Health Organization/International Agency for Research on Cancer: Lyon, France, 2002; Volume 82, pp. 1–590. [Google Scholar]
- Carballo, D.; Font, G.; Ferrer, E.; Berrada, H. Evaluation of mycotoxin residues on ready-to-eat food by chromatographic methods coupled to mass spectrometry in tandem. Toxins 2018, 10, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshannaq, A.; Yu, J.-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [Green Version]
- Peraica, M.; Radic, B.; Lucic, A.; Pavlovic, M. Toxic effects of mycotoxins in humans. Bull. World Health Organ. 1999, 77, 754–766. [Google Scholar] [PubMed]
- Probst, C.; Njapau, H.; Cotty, P.J. Outbreak of an acute aflatoxicosis in Kenya in 2004: Identification of the causal agent. Appl. Environ. Microbiol. 2007, 73, 2762–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, D.; Kang’ethe, E.; Lindahl, J.; Atherstone, C.; Wesonga, T. Aflatoxin: Impact on Animal Health and Productivity. Building an Aflatoxin Safe East African Community—Technical Policy Paper 4; IITA: Dar es Salam, Tanzania, 2015. [Google Scholar]
- Suleiman, E.A.; Elgabbar, M.A.; Khaliefa, K.A.; Omer, F.A. Aflatoxins in broiler chicks feed. Sudan J. Vet. Res. 2010, 25, 5–8. [Google Scholar]
- Banday, M.T.; Darzi, M.M.; Khan, A.A. Clinico-pathological and haemo-biochemical changes in broiler chicken following an outbreak of aflatoxicosis. Appl. Biol. Res. 2006, 8, 40–43. [Google Scholar]
- Rashid, N.; Bajwa, M.A.; Rafeeq, M.; Khan, M.A.; Ahmad, Z.; Tariq, M.M.; Wadood, A.; Abbas, F. Prevalence of aflatoxin B1 in finished commercial broiler feed from west central Pakistan. J. Anim. Plant. Sci. 2012, 22, 6–10. [Google Scholar]
- Akinmusire, O.O.; El-Yuguda, A.-D.; Musa, J.A.; Oyedele, O.A.; Sulyok, M.; Somorin, Y.M.; Ezekiel, C.N.; Krska, R. Mycotoxins in poultry feed and feed ingredients in Nigeria. Mycotoxin Res. 2019, 35, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Greco, M.V.; Franchi, M.L.; Golba, S.L.R.; Pardo, A.G.; Pose, G.N. Mycotoxins and mycotoxigenic fungi in poultry feed for food-producing animals. Sci. World J. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gathumbi, J.K.; Bebora, L.C.; Muchiri, D.J.; Ngatia, T.A. A survey of mycotoxins in poultry feeds used in Nairobi, Kenya. Bull. Anim. Health Prod. Afr. 1995, 43, 243–245. [Google Scholar]
- Nakavuma, J.; Kirabo, A.; Bogere, P.; Nabulime, M.M.; Kaaya, A.N.; Gnonlonfin, B. Awareness of mycotoxins and occurrence of aflatoxins in poultry feeds and feed ingredients in selected regions of Uganda. Int. J. Food Contam. 2020, 7, 1–10. [Google Scholar] [CrossRef]
- Kemboi, D.C.; Ochieng, P.E.; Antonissen, G.; Croubels, S.; Scippo, M.-L.; Okoth, S.; Kangethe, E.K.; Faas, J.; Doupovec, B.; Lindahl, J.F.; et al. Multi-mycotoxin occurrence in dairy cattle and poultry feeds and feed ingredients from Machakos town, Kenya. Toxins 2020, 12, 762. [Google Scholar] [CrossRef] [PubMed]
- FAO. Worldwide Regulations for Mycotoxins in Food and Feeds in 2003. FAO Food and Nutrition Paper 81. Available online: http://www.fao.org/3/y5499e/y5499e02.htm (accessed on 14 February 2021).
- FDA. Guidance for Industry: Action Levels for Poisonous or Deleterious Substances in Human Food and Animal Feed; US Department of Health and Human Services, US Food and Drugs Administration: Washington, DC, USA, 2017; Volume 2018. [Google Scholar]
- EU. Commission regulation (EU) No 165/2010 of 26 February 2010. Off. J. Eur. Union 2010, 50, 8–12. [Google Scholar]
- EC. Directive 2002/32/EC of the European parliament and of the council of 7 may 2002 on undesirable substances in animal feed. Off. J. Eur. Communities 2002, 140, 110–121. [Google Scholar]
- Tang, L.; Xu, L.; Afriyie-Gyawu, E.; Liu, W.; Wang, P.; Tang, Y.; Wang, Z.; Huebner, H.J.; Ankrah, N.A.; Ofori-Adjei, D.; et al. Aflatoxin-albumin adducts and correlation with decreased serum levels of vitamins A and E in an adult Ghanaian population. Food Addit. Contam. Part. A 2009, 26, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflatoxicosis in developing countries: A review of toxicology, exposure, potential health consequences and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar] [CrossRef] [PubMed]
- Amin, Y.A.; Mohamed, R.H.; Zakaria, A.M.; Wehrend, A.; Hussein, H.A. Effects of aflatoxins on some reproductive hormones and composition of buffalo’s milk. Comp. Clin. Pathol. 2019, 28, 1191–1196. [Google Scholar] [CrossRef]
- El Mahady, M.M.; Ahmed, K.A.; Badawy, S.A.; Ahmed, Y.F.; Aly, M.A. Pathological and hormonal effects of aflatoxins on reproduction of female albino rats. Middle East. J. Appl. Sci. 2015, 5, 998–1006. [Google Scholar]
- Umar, S.; Younus, M.; Rehman, M.U.; Aslam, A.; Shah, M.A.A.; Munir, T.; Hussain, S.; Iqbal, F.; Fiaz, M.; Ullah, S. Role of aflatoxin toxicity on transmissibility and pathogenicity of H9N2 avian influenza virus in turkeys. Avian Pathol. 2015, 44, 305–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trench, P.C.; Narrod, C.; Roy, D.; Tiongco, M. Responding to health risks along the value chain. 2020 Conference Paper 5. In Proceedings of the Leveraging Agriculture for Improving Nutrition and Health, New Delhi, India, 10–12 February 2011; pp. 1–54. [Google Scholar]
- Fouad, A.M.; Ruan, D.; El-Senousey, H.K.; Chen, W.; Jiang, S.; Zheng, C. Harmful effects and control strategies of aflatoxin B1 produced by Aspergillus flavus and Aspergillus parasiticus strains on Poultry: Review. Toxins 2019, 11, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andretta, I.; Kipper, M.; Lehnen, C.R.; Hauschild, L.; Vale, M.M.; Lovatto, P.A. Meta-analytical study of productive and nutritional interactions of mycotoxins in broilers. Poult. Sci. 2011, 90, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.M.; Ledoux, D.R.; Chester, L.F.B.; Samuels, C.A.N. A limited survey of aflatoxins in poultry feed and feed ingredients in Guyana. Vet. Sci. 2017, 2017, 60. [Google Scholar] [CrossRef] [Green Version]
- Bata-Vidács, I.; Kosztik, J.; Mörtl, M.; Székács, A.; Kukolya, J. Aflatoxin B1 and sterigmatocystin binding potential of non-Lactobacillus LAB Strains. Toxins 2020, 2020, 799. [Google Scholar] [CrossRef]
- Kosztik, J.; Mörtl, M.; Székács, A.; Kukolya, J.; Bata-Vidács, I. Aflatoxin B1 and sterigmatocystin binding potential of lactobacilli. Toxins 2020, 2020, 756. [Google Scholar] [CrossRef]
- Ozer, H.; Basegmez, H.O.; Whitaker, T.B.; Slate, A.B.; Giesbrecht, F.G. Sampling dried figs for aflatoxin—Part 1: Variability associated with sampling, sample preparation, and analysis. World Mycotoxin J. 2017, 10, 31–40. [Google Scholar] [CrossRef]
- Grace, D.; Lindahl, J.; Atherstone, C.; Kang’ethe, E.; Nelson, F.; Wesonga, T.; Manyong, V. Aflatoxin standards for feed. In Building an Aflatoxin Safe East African Community—Technical Policy Paper 7; IITA: Dar es Salam, Tanzania, 2015. [Google Scholar]
- Whitaker, T.B. Sampling foods for mycotoxins. Food Addit. Contam. 2006, 23, 50–61. [Google Scholar] [CrossRef]
- Matumba, L.; Whitaker, T.; Slate, A.; De Saeger, S. Current trends in sample size in mycotoxin in grains. Are we measuring accurately? Toxins 2017, 9, 276. [Google Scholar]
- Cheli, F.; Campagnoli, A.; Pinotti, P.; Fusi, E.; Dell’Orto, V. Sampling feed for mycotoxins: Acquiring knowledge from food. Ital. J. Anim. Sci. 2009, 8, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Food and Agriculture Organization of the United Nations. Mycotoxin Sampling Tool. User Guide. Version 1.0 (December 2013), Version 1.1 ed.; Food and Agriculture Organization of the United Nations: Rome, Italy, 2013; p. 62. [Google Scholar]
- Whitaker, T.B.; Slate, A.B. Comparing the USDA/AMS subsampling mill to a vertical cutter mixer type mill used to comminute shelled peanut samples for aflatoxin analysis. Peanut Sci. 2012, 39, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Kumphanda, J.; Matumba, L.; Whitaker, T.B.; Kasapila, W.; Sandahl, J. Maize meal slurry mixing: An economical recipe for precise aflatoxin quantitation. World Mycotoxin J. 2019, 12, 203–212. [Google Scholar] [CrossRef]
- Oulkar, D.; Goon, A.; Dhanshetty, M.; Khan, Z.; Satav, S.; Banerjee, K. High-sensitivity direct analysis of aflatoxins in peanuts and cereal matrices by ultra-performance liquid chromatography with fluorescence detection involving a large volume flow cell. J. Environ. Sci. Health Part B 2018, 53, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Okuma, T.A.; Huynh, T.P.; Hellberg, R. Use of Enzyme-linked immunosorbent assay to screen for aflatoxins, ochratoxin A, and deoxynivalenol in dry pet foods. Mycotoxin Res. 2018, 34, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taye, W.; Ayalew, A.; Chala, A.; Dejene, M. Aflatoxin B1 and total fumonisin contamination and their producing fungi in fresh and stored sorghum grain in East Hararghe, Ethiopia. Food Addit. Contam. Part B 2016, 9, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.T.T.; Nguyen, T.T.T.; Nguyen-Viet, H.; Tran, K.N.; Lindahl, J.; Randolph, D.G.; Ha, T.M.; Lee, H.S. Assessment of aflatoxin B1 in maize and awareness of aflatoxins in Son La, Vietnam. Infect. Ecol. Epidemiol. 2018, 8. [Google Scholar] [CrossRef]
- Huynh, T.P.; Ly, C. Recovery of aflatoxin B1 in a range of food commodities utilizing a matrix resistant ELISA. In Proceedings of the IAFP 2015 Annual Meeting, Portland, OR, USA, 25–28 July 2015. [Google Scholar]
- Wagner, C. Critical practicalities in sampling for mycotoxins in feed. J. Aoac Int. 2015, 98, 301–308. [Google Scholar] [CrossRef]
- Herrman, T. Sampling: Procedures for feed. MF-2036. Feed Manufacturing. Kansas State University Agricultural Experiment Station and Cooperative Extension Service: 2001. Department of Grain Science. Available online: http://www.oznet.ksu.edu/library/grsci2/MF2036 (accessed on 23 January 2014).
- Rao, T.N. Validation of Analytical Methods; IntechOpen: London, UK, 2018; Chapter 7. [Google Scholar] [CrossRef] [Green Version]
- BPU. Guidelines for Validation of Analytical Methods for Non-Agricultural Pesticide Active Ingredients and Products. Available online: www.hse.gov.uk/biocides/copr/pdfs/validation. (accessed on 14 October 2016).
- EC. Technical Material and Preparations: Guidance for Generating and Reporting Methods of Analysis in Support of Pre- and Post-Registration Data Requirements for Annex II (Part A, Section 4) and Annex III (Part A, Section 5) of Directive 91/414. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_ppp_app-proc_guide_phys-chem-ana_tech-mat-preps.pdf (accessed on 14 October 2016).
- OECD. Guidance document for single laboratory validation of quantitative analytical methods-guidance used in support of pre-and-post-registration data requirements for plant protection and biocidal products. In Environment, Health and Safety Publications Series on Testing and Assessment; No. 204 and Series on Biocides No. 9. ENV/JM/MONO (2014) 20; Organisation for Economic Co-operation and Development: Paris, France, 2014; Volume JT03360406. [Google Scholar]
- EC. Laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, 70, 12–33. [Google Scholar]
- EU. Commission recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding. Off. J. Eur. Union 2006, 229, 227–229. [Google Scholar]
- Dragacci, S.; Grosso, F. Validation of Analytical Methods to Determine the Content of Aflatoxin, Ochratoxin and Patulin in Foodstuffs of Vegetable Origin; European Commission BCR Information Chemical Analysis: Paris, France, 1999; pp. 1–40. [Google Scholar]
- Gerlach, R.W.; Dobb, D.E.; Raab, G.A.; Nocerino, J.M. Gy sampling theories in environmental studies. 1. Assessing soil splitting protocols. J. Chemom. 2002, 16, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.; Colwell, P.; Cowen, S.; Ellison, S.L.R.; Gray, K.; Elahi, S.; Farnell, P.; Slack, P.; Burns, D.T. Aflatoxins in Groundnuts–Assessment of the effectiveness of EU sampling and UK enforcement sample preparation procedures. J. Assoc. Public Anal. 2017, 45, 1–22. [Google Scholar]
- Food and Agriculture Organization of the United Nations. Sampling Plans for Aflatoxin Analysis in Peanuts and Corn. FAO Food and Nutrition Paper 55. Report of an FAO Technical Consultation Rome; Food and Agriculture Organization of the United Nations: Rome, Italy, 1993. [Google Scholar]
- Reiter, E.V.; Dutton, M.F.; Agus, A.; Nordkvist, E.; Mwanza, M.F.; Njobeh, P.B.; Prawano, D.; Haggblom, P.; Razzazi-Fazeli, E.; Zentek, J.; et al. Uncertainty from sampling in measurement of aflatoxins in animal feedstuffs: Application of the Eurachem/CITAC guidelines. Analyst 2011, 136, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Spanjer, M.C.; Scholten, J.M.; Kastrup, S.; Jorissen, U.; Schatzki, T.F.; Toyofuku, N. Sample comminution for mycotoxin analysis: Dry milling or slurry mixing? Food Addit. Contam. 2006, 23, 73–83. [Google Scholar] [CrossRef]
- IUPAC. Harmonized guidelines for single laboratory validation of methods of analysis. IUPAC Technical Report. Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- APVMA. Guidelines for the Validation of Analytical Methods for Active Constituent, Agricultural and Veterinary Chemical Products. Available online: http://www.apvma.gov.au (accessed on 14 October 2016).
- Shao, D.; Imerman, P.M.; Schrunk, D.E.; Ensley, S.M.; Rumbeiha, W.K. Intralaboratory development and evaluation of a high-performance liquid chromatography-fluorescence method for detection and quantitation of aflatoxins M1, B1, B2, G1, and G2 in animal liver. J. Vet. Diagn. Investig. 2016, 28, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Trucksess, M.W.; Weaver, C.M.; Oles, C.J.; Fry, F.S. Determination of aflatoxins B1, B2, G1, and G2 and ochratoxin A in ginseng and ginger by multitoxin immunoaffinity column cleanup and liquid chromatographic quantitation: Collaborative study. J. AOAC Int. 2008, 91, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Brera, C.; Debegnach, F.; Minardi, V.; Pannunzi, C.; Santis, B.D. Immunoaffinity column cleanup with liquid chromatography for determination of aflatoxin b1 in corn samples: Interlaboratory study. J. AOAC Int. 2007, 90, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.P.; Jackson, M.D.; Leek, J.M.; Samadpour, M. Sample preparation and analytical considerations for the US aflatoxin sampling program for shelled peanuts. Peanut Sci. 2018, 45, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Kang’ethe, E.K.; Gatwiri, M.; Sirma, A.J.; Ouko, E.O.; Mburugu-Mosoti, C.K.; Kitala, P.M.; Nduhiu, G.J.; Nderitu, J.G.; Mungatu, J.K.; Hietaniemi, V.; et al. Exposure of Kenyan population to aflatoxins in foods with special reference to Nandi and Makueni counties. Food Qual. Saf. 2017, 1, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.H.; Lewis, L.W.; Redwood, Y.A.; Kieszak, S.; Breiman, R.F.; Flanders, W.D.; Bell, C.; Mwihia, J.; Ogana, G.; Likimani, S.; et al. Comprehensive Assessment of Maize Aflatoxin Levels in Eastern Kenya, 2005–2007. Environ. Health Perspect. 2011, 119, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization/World Health Organization. Dietary exposure assessment of chemicals in food. In Environmental Health Criteria 240: Principles and Methods for the Risk Assessment of Chemicals in Food. A Joint Publication of the Food and Agriculture Organization of the United Nations and the World Health Organization. Chapter 6; Food and Agriculture Organization/World Health Organization: Geneva, Switzerland, 2009; pp. 1–95. [Google Scholar]
- EURO. GEMS/Food-EURO Second Workshop on Reliable evaluation of low-level contamination of food. Report on a workshop in the Frame of GEMS/Food-EURO. In Proceedings of the Workshop in the Frame of GEMS/Food-EURO, Kulmbach, Germany, 26–27 May 1995. [Google Scholar]
- Huong, B.T.M.; Tuyen, L.D.; Madsen, H.; Brimer, L.; Friis, H.; Dalsgaard, A. Total dietary intake and health risks associated with exposure to aflatoxin B1, ochratoxin A and fuminisins of children in Lao Cai Province, Vietnam. Toxins 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Ding, X.; Bai, Y.; Wu, L.; Yue, X.; Zhang, L. Risk Assessment and Prediction of Aflatoxin in Agro-Products; IntechOpen: London, UK, 2018; Chapter 18; pp. 319–334. [Google Scholar]
- Senerwa, D.M.; Sirma, A.J.; Mtimet, N.; Kang’ethe, E.K.; Grace, D.; Lindahl, J.F. Prevalence of aflatoxin in feeds and cow milk from five counties in Kenya. Afr. J. Food, Agric. Nutr. Dev. 2016, 16, 11004–11021. [Google Scholar] [CrossRef]
- Raad, F.; Nasreddine, L.; Hilan, C.; Bartosik, M.; Parent-Massin, D. Dietary exposure to aflatoxins, ochratoxin A and deoxynivalenol from a total diet study in an adult urban Lebanese population. Food Chem. Toxicol. 2014, 73, 35–43. [Google Scholar] [CrossRef]
- Bellio, A.; Daniela Manila Bianchi, D.M.; Gramaglia, M.; Loria, A.; Nucera, D.; Gallina, S.; Gili, M.; Decastelli, L. Aflatoxin M1 in Cow’s Milk: Method validation for milk sampled in Northern Italy. Toxins 2016, 8, 57. [Google Scholar] [CrossRef]
- Krska, R.; Richard, J.L.; Schuhmacher, R.; Slate, A.B.; Whitaker, T.B. Romer Labs Guide to Mycotoxins, 4th ed.; Anytime Publishing Services: Leicestershire, UK, 2012. [Google Scholar]
- Rodrigues, I.; Handl, J.; Binder, E.M. Mycotoxin occurrence in commodities, feeds and feed ingredients sourced in the Middle East and Africa. Food Addit. Contam. Part B 2011, 4, 168–179. [Google Scholar] [CrossRef] [Green Version]
- AOAC. Preparation of Standards for aflatoxins: Thin-layer chromatographic-Spectrometric methods (AOAC Official Method 970.44). In Official Methods of Analysis; Cunniff, P., Ed.; Health Protection Branch, AOAC International: Ottawa, ON, Canada, 2005; pp. 43–49. [Google Scholar]
- European Union Reference Laboratory. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; European Union Reference Laboratory: Lyngby, Denmark, 2016; pp. 1–52. [Google Scholar]
- Rossi, C.N.; Takabavashi, C.R.; Ono, M.A.; Saito, G.H.; Itano, E.N.; Kawamura, O.; Hirooka, E.Y.; Ono, E.Y.S. Immunoassay based on monoclonal antibody for aflatoxin detection in poultry feed. Food Chem. 2012, 132, 2211–2216. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.; Ly, C.; Knight, P.; Wolde-Mariam, W. Quantitation of aflatoxin B1 by ELISA in commodities that pose a matrix effect. In Proceedings of the AOAC 126th Annual Meeting, Las Vegas, Nevada, 30 September–3 October 2012. [Google Scholar]


{kind=link}
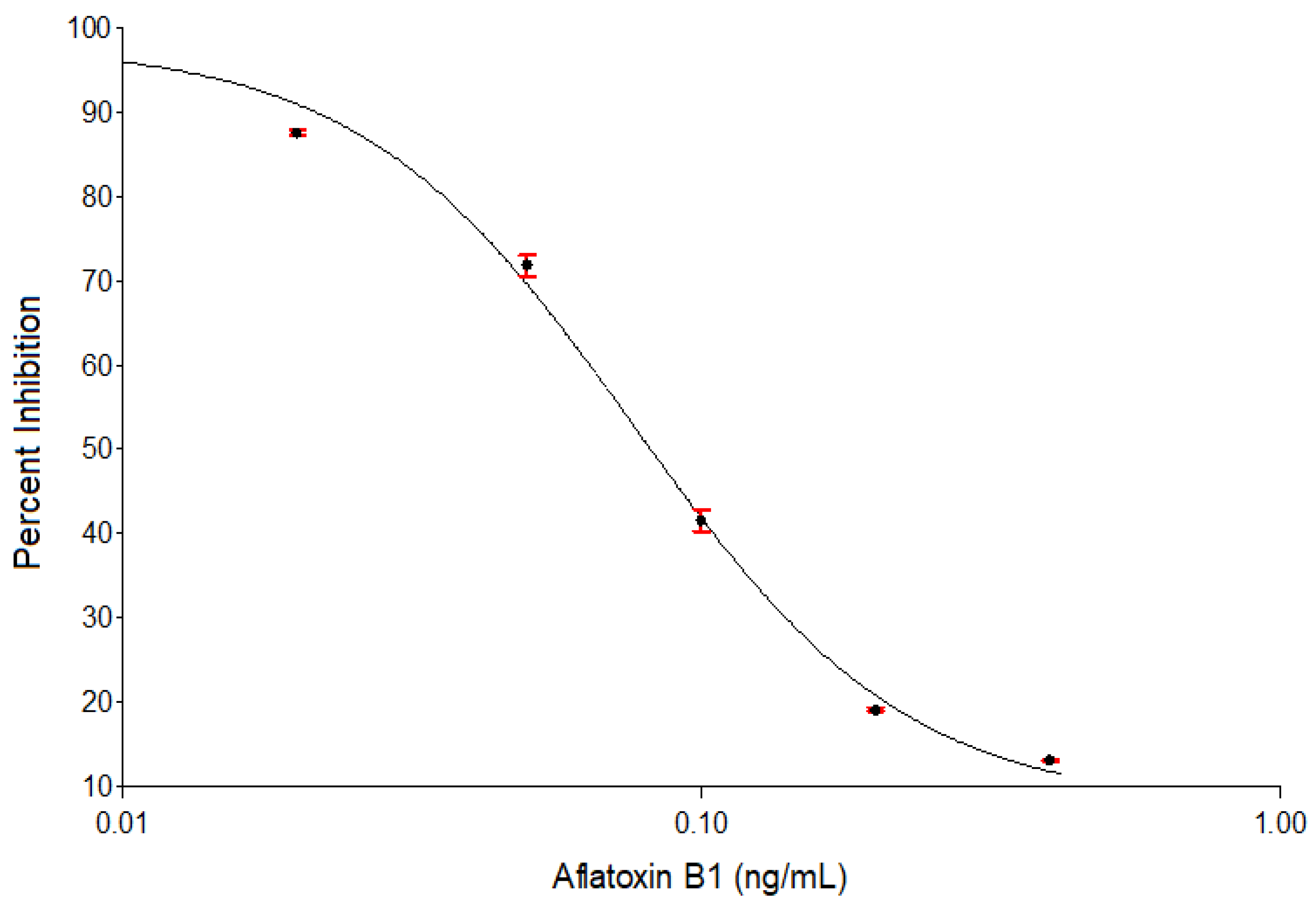{kind=link}
| Parameters Values of 4-Parameter Logistic Curve | |||||
|---|---|---|---|---|---|
| Maximum signal intensity (a) | Slope at inflection point (b) | Concentration at inflection point ((50% B/B0), IC50 (c) | Minimum signal intensity (d) | Coefficients of correlation (r) | Coefficients of determination (r2) |
| 98.9 | 1.58 | 0.072 ng/mL | 5.6 | 0.998 | 0.997 |
| Feed Aliquot Group | Extraction Conditions ε | Study |
|---|---|---|
| I (R = 6) * | (i) dry milling (ii) matrix/ACN a (25:133, w/v); (iii) ACN a extract/PBS-T b (10.6:989.4, v/v), (iv) final dilution factor = 500 | Spike and recovery (comparison of dry and wet milling) |
| II (R = 6) * | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (2:133, w/v); (iii) ACN a extract/PBS-T b (100:650, v/v), (iv) final dilution factor = 1247 | |
| III (R = 10) * | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (1.3:130, w/v); (iii) ACN a extract/PBS-T b (100:900, v/v), (iv) final dilution factor = 2500 | Spike and recovery studies (comparison of three extraction conditions used in wet milling) |
| IV (R = 10) * | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (1.3:65, w/v); (iii) ACN a extract/PBS-T b (100:900, v/v), (iv) final dilution factor = 1250 | |
| V (R = 10) * | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (1.3:86.5, w/v); (iii) ACN a extract/PBS-T b (100:650, v/v), (iv) final dilution factor = 1247 | |
| VI (R = 10) | (i) dry milling (ii) matrix/ACN a (25:130, w/v); (iii) ACN a extract/PBS-T b (100:900, v/v), (iv) final dilution factor = 52 | Intra-laboratory variability (Repeatability) |
| VII (R = 10) | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (1.3:130, w/v); (iii) ACN a extract/PBS-T b (100:900, v/v), (iv) final dilution factor = 2500 | |
| VIII (R = 10) | (i) dry milling (ii) matrix/ACN a (1.3:130, w/v); (iii) ACN a extract/PBS-T b (100:900, v/v), (iv) final dilution factor = 1000 | |
| IX (R = 10) | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (1.3:130, w/v); (iii) ACN a extract/PBS-T b (100:900, v/v) (iv) final dilution factor = 2500 | |
| X (R = 15) | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry n/ACN a, (1.3:130, w/v); (iii) ACN a extract: PBS-T b (100:900, v/v); (iv) final dilution factor = 2500 | Intermediate variability (within-laboratory reproducibility) |
| XI (R = 15) | (i) wet milling @ matrix/water (25:37.5, w/w); (ii) slurry/ACN a, (1.3:86.5, w/v); (iii) ACN a extract/PBS-T b (100:650, v/v) (iv) Final dilution factor = 1247 | |
| XII (R = 4) | (i) dry milling (ii) ACN a extract/PBS-T b (1:1000, v/v) (iii) final dilution factor = 1000 | Determination of LOD and LOQ |
| XIII (R = 5) | (i) dry milling ACN a extract/ACN a/PBS-T b (1:5:100, v/v/v) (ii) final dilution factor = 500 | |
| XIV (R = 5) | (i) wet milling (ii) ACN a extract/PBS-T b (1:1247, v/v) (iii) final dilution factor = 1247 |
| Milling Method | Group | Aflatoxin Extraction Conditions | * Mean AFB1 % Recovery | % Bias | Measurement Variability | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expected Concentration | Size of Analytical Sample | DF of Water Slurry | DF in ACN * | DF in PBS-T b | Final DF | StDev. | CV | CV Effect | HorRat ** Value | ||||
| Dry | I (R = 6) | 20 ppb (R = 3) | 25 g | - | 5.3 | 94.3 | 500 | 57 | ±6 | 10.9 | 0.57 | ||
| 100 ppb (R = 3) | 103 | ±13 | 12.4 | 0.82 | |||||||||
| All replicates | 80 a | −20 | ±26 | 33.1 | 9.5 R4 | 0.57–0.82 | |||||||
| Wet | II (R = 6) | 20 ppb (R = 3) | 2 g | 2.5 | 66.5 | 7.5 | 1247 | 119 | ±25 | 21.2 | 0.95 | ||
| 100 ppb (R = 3) | 123 | ±9 | 7.4 | 0.8 | |||||||||
| All replicates | 121 a | 21 | ±17 | 14.1 h | −9.5 R2 | 0.8–0.95 | |||||||
| Wet | III (R = 10) | 20 ppb (R = 5) | 1.3 g | 2.5 | 100 | 10 | 2500 | 145 | ±19 | 12.8 | 0.58 | ||
| 100 ppb (R = 5) | 112 | ±9 | 17.4 | 0.44 | |||||||||
| All replicates | 129 b,c | 29 | ±22 | 17.2 | −3.8 R3 | 0.44–0.58 | |||||||
| IV (R = 10) | 20 ppb (R = 5) | 1.3 g | 2.5 | 50 | 10 | 1250 | 49 | ±12 | 23.2 | 1.04 | |||
| 100 ppb (R = 5) | 85 | ±15 | 18.1 | 1.04 | |||||||||
| All replicates | 67 b | −33 | ±23 | 34.2 | 13.3 R5 | 1.04 | |||||||
| V (R = 10) | 20 ppb (R = 5) | 1.3 g | 2.5 | 66.5 | 7.5 | 1247 | 80 | ±13 | 15.8 | 0.71 | |||
| 100 ppb (R = 5) | 80 | ±6 | 6.9 | 0.39 | |||||||||
| All replicates | 80 c | −20 | ±9 | 11.5 | −9.5 R1 | 0.39–0.71 | |||||||
| Groups II and V replicates analyzed together (R = 16) | 20 ppb (R = 8) | - | 2.5 | 66.5 | 7.5 | 1247 | 94 | ±26 | 27.6 | 1.24 | |||
| 100 ppb (R = 8) | 96 | ±23 | 24.1 | 1.38 | |||||||||
| All replicates | 95 | −5 | ±24 | 25.0 | 1.24–1.38 | ||||||||
| Sampling Stage | Intra-Laboratory Assay Precision (Within-Laboratory Repeatability) | Intermediate Precision (Within-Laboratory Reproducibility) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group Number/ Mass Reduction Method | Milling Method and Size of Analytical Sample | EC | AFB1 Level (ppb) Mean ± Sd. | Variability | Group Number/ Mass Reduction Method | Milling Method and Size of Analytical Sample | EC | AFB1 Level (ppb) Mean ± Sd | Variability | |||
| RSDr | HorRat ValueEffect | RSDR | HorRat Value Effect | |||||||||
| Secondary | Group VI Coning and quartering (R = 10) | Dry 25 g | 1 | 15 ± 1 | 6.5 | 0.3 H1 | - | |||||
| Tertiary | Group VII Water slurry (R = 10) | Wet 1.3 g | 2 | 68 ± 17 | 24.5 | 1.5 H5 | Group X Water slurry (R = 15) | Wet 1.3 g | 2 | 69 ± 21 | 30.3 | 1.3 H4 |
| Tertiary | Group VIII Coning and quartering (R = 10) | Dry 1.3 g | 3 | 73 ± 67 | 91.5 | 5.8 H6 | Group XI Water slurry (R = 15) | Wet 1.3 g | 4 | 19 ± 5 | 26.9 | 0.9 H2 |
| Tertiary | Group IX Water slurry (R = 10) | Wet 1.3 g | 2 | 50 ± 10 | 20.6 | 1.2 H3 | - | |||||
| Baseline Response (OD) Statistics | Limits of Detection and Quantification in µg AFB1 Per kg of Chicken Feed | ||
|---|---|---|---|
| Dry Milling | Wet Milling | ||
| Group XII (R = 4) (Final Dilution Factor = 1000) | Group XIII (R = 5) (Final Dilution Factor = 500) | Group XIV (R = 5) (Final Dilution Factor = 1247) | |
| Mean-2StDev (Limit of detection) | 16.3 | 10.6 | 7.5 |
| Mean-5StDev (Limit of quantification) | 31 | 22.3 | 16 |
| Baseline response (OD) statistics | Magnitude of signal, noise, their ratio and precision (absorbance) | ||
| Group XII | Group XIII | Group XIV | |
| B0 | 1.773 | 1.752 | 1.871 |
| StDev (blank) | 0.0399 | 0.0485 | 0.0339 |
| Signal/noise ratio | 44.44 | 36.12 | 55.19 |
| Coefficient of variance | 2.34 | 2.99 | 1.97 |
| Concentration and Optical Density of AFB1 Standard Solutions | ||||||
|---|---|---|---|---|---|---|
| Solution 1 | Solution 2 | Solution 3 | Solution 4 | Solution 5 | Solution 6 | |
| Spiked AFB1 level (ng/mL) | 0.02 | 0.05 | 0.1 | 0.2 | 0.4 | 0.5 |
| Measured level (ng/mL) | 0.03 | 0.06 | 0.12 | 0.28 | 0.36 | >0.4 |
| Measured optical density | 1.429 | 1.106 | 0.663 | 0.297 | 0.226 | 0.199 |
| Number of Replicates Required to Attain Acceptable Intra-Assay Precision | |||||
|---|---|---|---|---|---|
| Two Replicates Required | Three Replicates Required | Four Replicates Required | Five Replicates Required | Six Replicates Required | |
| Number and percentage of samples analyzed | 188 (74.9%) | 51 (20.3%) | 8 (3.2%) | 2 (0.8%) | 2 (0.8%) |
| Study Reference | Characteristics of Aflatoxin Test Procedures | ||||
|---|---|---|---|---|---|
| Homogenization Method | Aggregate Sample (kg) | Laboratory Sample (kg) | Size of Test Portion | Analytical Method | |
| [15] | Dry milling | 4 | Not given | 5 g | LC-MS/MS |
| [16] | Dry milling | 1–2 | 1 | 5 g | ELISA |
| [19] | Dry milling | 1 | 1 | 5 g | LC-MS/MS |
| [32] | Dry milling | Not given | Not given | Not given | HPLC/ELISA |
| [18] | Dry milling | Not given | Not given | 50 g | VICAM Fluorimeter |
| [13] | Not given | Not given | Not given | Not given | Not given |
| [14] | Not given | 1 | Not given | Not given | TLC |
| Novel method | Wet milling (water slurring) | >2 | 2 | at least 25 g × 2 | ELISA or any other quantification method |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibugu, J.; Mdachi, R.; Munga, L.; Mburu, D.; Whitaker, T.; Huynh, T.P.; Grace, D.; Lindahl, J.F. Improved Sample Selection and Preparation Methods for Sampling Plans Used to Facilitate Rapid and Reliable Estimation of Aflatoxin in Chicken Feed. Toxins 2021, 13, 216. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13030216
Kibugu J, Mdachi R, Munga L, Mburu D, Whitaker T, Huynh TP, Grace D, Lindahl JF. Improved Sample Selection and Preparation Methods for Sampling Plans Used to Facilitate Rapid and Reliable Estimation of Aflatoxin in Chicken Feed. Toxins. 2021; 13(3):216. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13030216
Chicago/Turabian StyleKibugu, James, Raymond Mdachi, Leonard Munga, David Mburu, Thomas Whitaker, Thu P. Huynh, Delia Grace, and Johanna F. Lindahl. 2021. "Improved Sample Selection and Preparation Methods for Sampling Plans Used to Facilitate Rapid and Reliable Estimation of Aflatoxin in Chicken Feed" Toxins 13, no. 3: 216. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13030216