The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium


1
A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch, Russian Academy of Sciences, 690041 Vladivostok, Russia
2
Marine Animal Ecology Group, Department of Animal Sciences, Wageningen University and Research, P.O. Box 338, 6700 AH Wageningen, The Netherlands
*
Author to whom correspondence should be addressed.
Toxins 2021, 13(6), 410; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060410
Submission received: 19 May 2021
/
Revised: 31 May 2021
/
Accepted: 7 June 2021
/
Published: 9 June 2021
(This article belongs to the Special Issue Analysis and Evaluation of Tetrodotoxin)
Abstract
:Tetrodotoxin (TTX)-producing bacteria have attracted great interest as a model system for study of the TTX biosynthetic route. Here, we report the complete genome of the TTX-producing bacterium Bacillus sp. 1839. The genome of the strain Bacillus sp. 1839, previously isolated from the TTX-bearing marine ribbon worm Cephalothrix cf. simula, was obtained using second generation Illumina and third generation nanopore sequencing technologies. Phylogenetic analysis has classified this strain as Cytobacillus gottheilii.
Key Contribution: This is the first report on the genome of the tetrodotoxin-producing bacterium.
1. Introduction
Tetrodotoxin (TTX), broadly distributed in marine ecosystems, is one of the most studied neurotoxins of the 20th–21st centuries [1]. Its ability to selectively bind voltage-gated sodium channels resulted in the popularity of the toxin in medical, pharmaceutical, and scientific spheres [2]. Despite the wide distribution, the molecular basis of TTX biosynthesis is still unresolved. The first attempt to decipher the genes involved in TTX production was made by Liu et al. [3]. The authors suggested the TTX-producing ability of the bacteria Aeromonas sp. strain Ne-1 was associated with the copy number of plasmid pNe-1, containing 32 open reading frames encoding hypothetical proteins. However, the bacteria lost the plasmid after 18 h of culture. In the other work, the authors suggested an association between some natural product biosynthesis genes (polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS)) and the TTX-producing ability of the microflora of toxic gastropods [4].
Bacillus sp. 1839 was isolated from the TTX-bearing nemertean Cephalothrix cf. simula in 2014 [5]. Confocal laser scanning microscopy with polyclonal antibodies against TTX allowed us to reveal TTX-positive labeling in the cells of the strain. Further detailed investigations with immunoelectron microscopy with anti-TTX antibodies revealed that toxin labeling was directly linked with the sporulation forms of the bacterium [6]. The life cycle and sporulation conditions studies [7,8] showed that TTX labeling was preserved through numerous passages for several years after the discovery of the strain. In 2019, the TTX producing ability of the strain was confirmed with high-performance liquid chromatography with tandem mass spectrometry [9]. TTX was revealed in the culture of the strain enriched with spores, confirming previous results.
A bacterial strain with TTX production in laboratory conditions is of great interest for the toxin biogenesis investigation. This study is the first to present the complete genome of a bacterium with in vitro TTX producing ability.
2. Results
2.1. General Genome Features of Bacillus sp. 1839
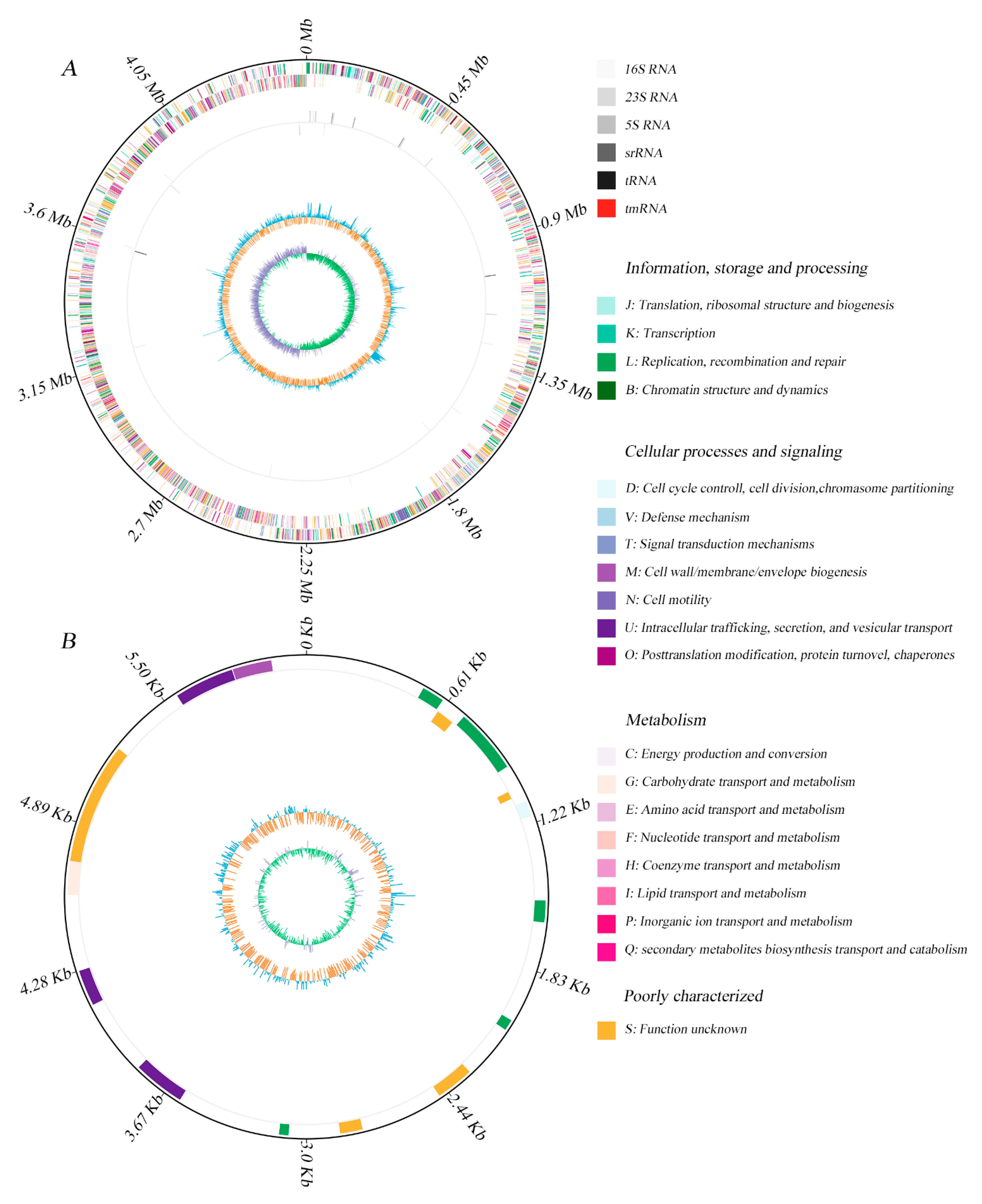
The specific features of the Bacillus sp. 1839 genome are summarized in Table 1 and Figure 1. The genome of the strain consists of a single 4.5 Mb circular chromosome with 39% GC content (Figure 1A) and a 0.06 Mb plasmid with 34% GC content (Figure 1B). The Bacillus sp. 1839 genome is predicted to include 4527 total genes, of which 4369 (96.5%) are protein-coding genes, 119 (2.6%) are RNA-coding, and 39 (0.9%) are pseudogenes (Table 1). Among the predicted genes, 3508 (77.5%) are associated with general Clusters of Orthologous Groups (COG) function categories (Table 2), however, 22.8% of them are poorly characterized and are assigned to the S group with unknown functions. Among all COG groups, genes encoding transcription (K, 6.2%), amino acid transport and metabolism (E, 5.8%), inorganic ion transport and metabolism (P, 5.7%), and carbohydrate transport and metabolism (G, 4.9%) are the most abundant. Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, 2314 coding sequences (CDSs) of Bacillus sp. 1839 genome were assigned to 212 KEGG pathways (Supplementary Table S1). Among all the KEGG pathways, “Metabolic pathways” (547), “Biosynthesis of secondary metabolites” (254), “Microbial metabolism in diverse environments” (129), “Biosynthesis of amino acids” (104), “Biosynthesis of cofactors” (112), “Two-component system in signal transduction” (91), “ABC transporters” (90), and “Carbon metabolism” (81) accounted for the largest proportion. Comparative genomic analysis of Bacillus sp. 1839 using antiSMASH identified four gene clusters related to secondary metabolite production (Table 3).
Mobile genetic elements predicted in the genome of Bacillus sp. 1839 are summarized in Supplementary Table S2. Analysis of transposable elements revealed numerous insertion sequences (IS) distributed over the genome of the strain. The majority of IS belong to the IS1182 (76), followed by IS3 (10), IS21 (3), IS4 (1), IS110 (1), and IS1595 (1) families. Eight genomic islands (GEIs) are found in the genome with the IslandViewer4. The largest GEI is also assigned as an intact prophage by the PHASTER server. A total of two intact (score >90), one incomplete (score <70), and one questionable (score 70–90) prophage regions were predicted in the genome of the strain, indicating previous phage infection. Two clustered regularly interspaced short palindromic repeats (CRISPR) loci were also detected.
2.2. Phylogenetic Analysis and Genome Similarity Measures
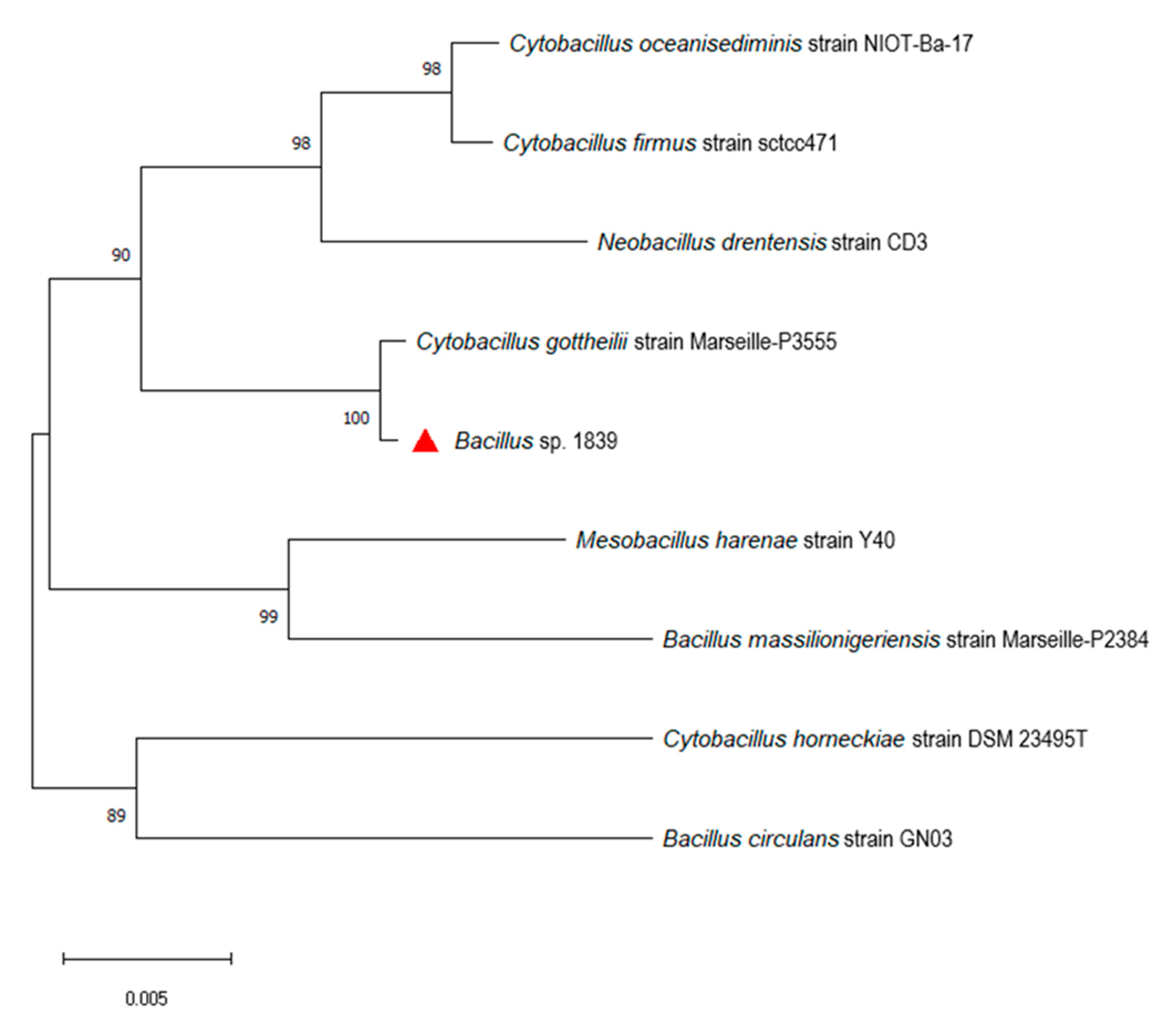
The phylogenetic tree based on 16S rRNA gene sequences revealed that Bacillus sp. 1839 is closest to Cytobacillus gottheilii (Figure 2). Up to now, only two complete genome sequences of this species have been deposited to the National Center for Biotechnology Information (NCBI) database (Table 4). We used these genomes for a more detailed analysis. As shown in Table 5, the closely related Cytobacillus strains resulted in a high average nucleotide identity (ANI) (>97%), exceeding the threshold value of 95% for distinguishing different species. Digital DNA–DNA hybridization (dDDH) values of Bacillus sp. 1839 against reference genomes were in the range of 79.6–85.9%, also exceeding the 70% DDH cutoff for species delineation (Table 6). This combination of analyses allowed us to classify Bacillus sp. 1839 as Cytobacillus gottheilii.
3. Discussion
In this study, we have described the genomic and phylogenetic features of the TTX-producing bacterium Bacillus sp. 1839. The phylogenetic analysis allowed to assign this strain to Cytobacillus gottheilii. The strains of this species were not earlier reported in TTX-production. Due to the complex structure of TTX, its biosynthetic pathway is not even predicted to date. It is assumed that the carbon backbone of TTX may originate through a polyketide, C5 branched sugar, or terpene [10]. The specific guanidinium moiety of the toxin can be obtained from a donor, such as an arginine, via amidinotransferase, similarly to amidino group transfer from l-arginine in saxitoxin biosynthesis [11], or NRPS and PKS systems [10]. The KEGG pathway database used to map the Bacillus sp. 1839 genome assigned 15 CDSs to “Arginine biosynthesis” and 17 CDSs to “Arginine and proline metabolism” indicating the potential ability of the bacterium to involve arginine in TTX production. Terpene and PKS gene clusters were also mined in the genome of the strain using antiSMASH. This study is the first to show the genome of a bacterial strain capable of TTX production in the laboratory—a good candidate for the unraveling of the molecular mechanisms of TTX synthesis. Genome sequencing is the first step allowing further experimental work aimed at gene cloning and expression, and reconstruction of the TTX synthetic and metabolic networks. The genome description and taxonomic classification opens the door to the comparative study of mutational patterns, ecological adaptations, and virulence determinants of the TTX producer and its closely related bacterial strains. Moreover, the genome obtained gives the possibility to reveal genes and traits not present in the other representatives of this species or, on the contrary, to find common features indicating their TTX-producing ability. Further investigations with the strain will be focused on the transcriptome studies on different stages of the life cycle of the bacterium.
4. Materials and Methods
4.1. DNA Extraction
The strain Bacillus sp. 1839 (KF444411-KF444416) was previously isolated from the TTX-bearing nemertean Cephalotrix cf. simula (Ivata, 1952) [5]. For DNA analysis, a strain was obtained from the Collection of Marine Heterotrophic Bacteria, A.V. Zhirmunsky National Scientific Centre of Marine Biology, Far Eastern Branch of the Russian Academy of Sciences. The strain was aerobically cultivated in 2 mL of Youschimizu–Kimura liquid medium [5] at 23 °C overnight. Bacteria were centrifuged at 3000 g for 10 min. The bacterial pellet was suspended in 1 mL of lysis buffer containing 20 мM Tris-HCL (pH 8.0), 2 мM EDTA, 1.2% Triton X-100, and 20 mg/mL lysozyme, and incubated for 30 min at 37 °C. Genomic DNA of the strain was extracted using a GeneJET Genomic DNA Purification Kit #K0721 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. For plasmid DNA extraction through Illumina sequencing, a GeneJET Plasmid Miniprep Kit # K0502 (Thermo Fisher Scientific, Waltham, MA, USA) was used. The DNA quality was evaluated using 1% agarose gel electrophoresis, an UV5Nano spectrophotometer (Mettler Toledo, Columbus, OH, USA), and a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The optical density ratio at 260/280 nm and 260/230 nm was >1.8 and >2.0, respectively. DNA was stored at −20 °C until further processing.
4.2. Genome Sequencing
The complete genome of strain Bacillus sp. 1839 was sequenced by the Illumina HiSeq 2500 (Illumina Inc., San Diego, CA, USA) and MinIon (Oxford Nanopore Technologies, Oxford, UK) platforms. Sequencing on the Illumina HiSeq 2500 system was performed at Genoanalytica Company (Moscow, Russia). Genome DNA was fragmented by the Covaris M220 sonicator (Covaris, Woburn, MA, USA). Libraries were constructed using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (Illumina Inc., San Diego, CA, USA) for paired-end sequencing, with a target average insert size of 200 bp. For nanopore sequencing, genomic DNA was fragmented by passing through a small gauge needle. The fragmented genomic DNA was used to construct the library with the 1D Genomic DNA by ligation kit SQK-LSK108 following the instructions provided by the manufacturer. Sequencing was carried out on a MinION Mk1B sequencer (Oxford Nanopore Technologies) using an R9.4.1 Flow Cell. The read qualities were examined by FastQC.
4.3. Genome Assembly
Low quality reads from the Illumina HiSeq 2500 system were filtered out before de novo genome assembly by a St. Petersburg genome assembler (SPAdes) v. 3.7.1. [12] (kmer = 127), followed by genome finishing using the CONTIGuator tool v. 2.7 3. [13]. The filtered Illumina reads were used to improve the de novo assembly from the MinIon reads. The reads from the MinIon system were assembled by the Staden Package pipeline v. 2.0.0b11 (http://staden.sourceforge.net/ (accessed on 6 June 2021)). The final contigs were circularized by Unicycler v. 0.4.8 [14] and validated by BUSCO [15]. The final assembly was 98.3% complete with 0.4% of the sequence predicted to be missing, as estimated by BUSCO.
4.4. Genome Annotation
The complete genome sequence of Bacillus sp. 1839 was annotated using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP). The gene functions were determined against the NCBI UniProt/Swiss-Prot and non-redundant (NR) protein databases, COG, Gene Ontology (GO), and KEGG databases with the E-value cutoff set to 10−5 and subsequent filtering for the best hit. tRNA and rRNA were identified by tRNAscan-SE v. 2.0 [16] and RNAmmer 1.2 [17], respectively. Circular representation of the genome including noncoding RNAs and gene function annotations was generated using the Circos software (version v.0.69-9) (http://www.circos.ca/ (accessed on 6 June 2021)). IS were predicted and classified with the ISFinder platform [18] against the ISfinder database v. 2.0 (http://www-is.biotoul.fr (accessed on 6 June 2021)). GEIs were detected with the IslandViewer4 online server using IslandPick, SIGI-HMM, and IslandPath-DIMOB prediction methods with default parameters [19]. CRISPR loci were detected using the CRISPRCasFinder online server [20]. Prophages in the genome were predicted with PHASTER online server [21,22]. The potential secondary metabolic gene clusters were predicted using antiSMASH v. 5.0 [23].
4.5. Phylogenetic Analysis and Genome Similarity Calculations
The 16S rRNA gene sequences of some strains closely related to Bacillus sp. 1839 were obtained by the BLASTN search against the NCBI database. The phylogenetic tree of Bacillus sp. 1839 and closely related species based on the 16S rRNA gene sequences was constructed using the neighbor-joining method [24] in MEGA X [25]. The evolutionary distances were computed using the maximum composite likelihood method [26] with 1000 bootstrap replications [27]. For genome similarity analysis, the genomes of two organisms closely related to the newly sequenced Bacillus sp. 1839 species were retrieved from the NCBI GenBank database (Table 4). The ANI values between the Bacillus sp. 1839 and closely related species Cytobacillus spp. were calculated using the BLASTALL algorithm (ANIb) and tetranucleotide frequency correlation coefficient (Tetra) with default parameters of the web server JSpecies v 1.2.1 [28]. Pairwise dDDH values were calculated using the Genome-to-Genome Distance Calculator (GGDC 2.1) [29].
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/toxins13060410/s1, Table S1: KEGG categories of Bacillus sp. 1839 genome, Table S2: Mobile genetic elements in Bacillus sp. 1839 genome.
Author Contributions
D.I.M., R.N. and T.Y.M. designed the project. D.I.M. performed the bacterial strain cultivation and DNA isolation, analyzed the data, and wrote the manuscript. R.N. performed the genome sequencing on the MinIon platform. T.Y.M. performed sequence processing, genome assembly and annotation, and phylogenetic analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The complete genome sequence of Bacillus sp. 1839 has been deposited in GenBank under the BioProject PRJNA707608 (accession number SRX10299645-SRX10299647). The annotated genome and plasmid are available in GenBank under accession numbers CP071709-CP071710.
Conflicts of Interest
The authors declare no competing interests.
References
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [Green Version]
- Melnikova, D.I.; Khotimchenko, Y.S.; Magarlamov, T.Y. Addressing the issue of tetrodotoxin targeting. Mar. Drugs 2018, 16, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wei, F.; Lu, Y.; Ma, T.; Zhao, J.; Gong, X.; Bao, B. Production level of tetrodotoxin in Aeromonas is associated with the copy number of a plasmid. Toxicon 2015, 101, 27–34. [Google Scholar] [CrossRef]
- Pratheepa, V.; Alex, A.; Silva, M.; Vasconcelos, V. Bacterial diversity and tetrodotoxin analysis in the viscera of the gastropods from Portuguese coast. Toxicon 2016, 119, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Beleneva, I.A.; Magarlamov, T.Y.; Kukhlevskii, A.D. Characterization, identification, and screening for tetrodotoxin production by bacteria associated with the Cephalotrix simula (Ivata, 1952) proboscis worm. Mikrobiologiia 2014, 83, 312–319. [Google Scholar] [CrossRef]
- Magarlamov, T.Y.; Beleneva, I.A.; Chernyshev, A.V.; Kuhlevsky, A.D. Tetrodotoxin-producing Bacillus sp. from the ribbon worm (Nemertea) Cephalothrix simula (Iwata, 1952). Toxicon 2014, 85, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Shokur, O.A.; Magarlamov, T.Y.; Melnikova, D.I.; Gorobets, E.A.; Beleneva, I.A. Life cycle of tetrodotoxin producing Bacillus sp. on solid and liquid medium: Light and electron microscopy studies. Russ. J. Mar. Biol. 2016, 42, 252–257. [Google Scholar] [CrossRef]
- Magarlamov, T.Y.; Melnikova, D.I.; Shokur, O.A.; Gorobets, E.A. Rapid production of tetrodotoxin-like compounds during sporulation in a marine isolate Bacillus sp. 1839. Microbiology 2017, 86, 192–196. [Google Scholar] [CrossRef]
- Melnikova, D.I.; Vlasenko, A.E.; Magarlamov, T.Y. Stable tetrodotoxin production by Bacillus sp. strain 1839. Mar. Drugs 2019, 17, 704. [Google Scholar] [CrossRef] [Green Version]
- Chau, R.; Kalaitzis, J.A.; Neilan, B.A. On the origins and biosynthesis of tetrodotoxin. Aquat. Toxicol. 2011, 104, 61–72. [Google Scholar] [CrossRef]
- Kellmann, R.; Mihali, T.K.; Jeon, Y.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling genomes and mini-metagenomes from highly chimeric reads. J. Comput. Biol. 2013, 10, 714–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galardini, M.; Biondi, E.G.; Bazzicalupo, M.; Mengoni, A. CONTIGuator: A bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol. Med. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucl. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNammer: Consistent annotation of ribosomal RNA genes. Nucl. Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Pérochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucl. Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucl. Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucl. Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucl. Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucl. Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucl. Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Graphic circular map of Bacillus sp. 1839 chromosome (A), and plasmid (B). From outer to inner rings, circle 1: the size of the complete genome; circle 2: the predicted protein-coding genes on the sense and antisense strands (colored according to Clusters of Orthologous Groups categories); circle 3: RNA genes; circle 4: G + C content, with >39% G + C in blue, with ≤39% G + C in orange; circle 5: G + C skew, with G% > C% in green, with G% < C% in violet.
Figure 1.
Graphic circular map of Bacillus sp. 1839 chromosome (A), and plasmid (B). From outer to inner rings, circle 1: the size of the complete genome; circle 2: the predicted protein-coding genes on the sense and antisense strands (colored according to Clusters of Orthologous Groups categories); circle 3: RNA genes; circle 4: G + C content, with >39% G + C in blue, with ≤39% G + C in orange; circle 5: G + C skew, with G% > C% in green, with G% < C% in violet.

Figure 2.
Neighbor-joining phylogenetic tree of Bacillus sp. 1839 and other closely related strains based on 16S rRNA gene sequences. Evolutionary distances are computed by the maximum likelihood method with 1000 bootstrap replications. Scale bar indicates 0.005 nucleotide substitutions per site. Bacillus sp. 1839 is indicated by the red triangle.
Figure 2.
Neighbor-joining phylogenetic tree of Bacillus sp. 1839 and other closely related strains based on 16S rRNA gene sequences. Evolutionary distances are computed by the maximum likelihood method with 1000 bootstrap replications. Scale bar indicates 0.005 nucleotide substitutions per site. Bacillus sp. 1839 is indicated by the red triangle.


{kind=link}
{kind=link}
Table 1.
The general genome features of Bacillus sp. 1839.
| Feature | Value | |
|---|---|---|
| Chromosome | Plasmid | |
| Genome size (bp) | 4,523,455 | 61,233 |
| G + C content (mol %) | 39 | 34 |
| Genes number (total) | 4458 | 69 |
| Coding sequences total number | 4339 | 69 |
| Coding sequences with protein number | 4300 | 69 |
| Protein-coding genes number | 4300 | 69 |
| rRNAs number | 11, 10, 10 (5S, 16S, 23S) | 0 |
| tRNAs number | 83 | 0 |
| ncRNAs number | 5 | 0 |
| Pseudogenes number | 39 | 0 |
| Insertion sequences number | 92 | 0 |
| Genomic islands number | 9 | 0 |
| Clustered regularly interspaced short palindromic repeats number | 2 | 0 |
| Prophage number | 4 | 0 |
Table 2.
Genes associated with general Clusters of Orthologous Groups (COG) functional categories of Bacillus sp. 1839.
Table 2.
Genes associated with general Clusters of Orthologous Groups (COG) functional categories of Bacillus sp. 1839.
| Code | Value | % 1 | Function Description |
|---|---|---|---|
| INFORMATION STORAGE AND PROCESSING | |||
| B | 1 | 0.02 | Chromatin structure and dynamics |
| J | 171 | 3.9 | Translation, ribosomal structure, and biogenesis |
| K | 269 | 6.2 | Transcription |
| L | 197 | 4.5 | Replication, recombination, and repair |
| METABOLISM | |||
| C | 189 | 4.3 | Energy production and conversion |
| E | 255 | 5.8 | Amino acid transport and metabolism |
| F | 88 | 2 | Nucleotide transport and metabolism |
| G | 213 | 4.9 | Carbohydrate transport and metabolism |
| H | 100 | 2.3 | Coenzyme transport and metabolism |
| I | 88 | 2 | Lipid transport and metabolism |
| P | 248 | 5.7 | Inorganic ion transport and metabolism |
| Q | 29 | 0.7 | Secondary metabolites biosynthesis, transport, and catabolism |
| CELLULAR PROCESSES AND SIGNALING | |||
| D | 36 | 0,8 | Cell cycle control, Cell division, and chromosome partitioning |
| M | 170 | 3.9 | Cell wall/membrane/envelope biogenesis |
| N | 43 | 1 | Cell motility |
| O | 113 | 2.6 | Posttranslational modification, protein turnover, chaperones |
| T | 191 | 4.4 | Signal transduction mechanisms |
| U | 38 | 0.9 | Intracellular trafficking, secretion, and vesicular transport |
| V | 75 | 1.7 | Defense mechanisms |
| POORLY CHARACTERIZED | |||
| S | 994 | 22.8 | Function unknown |
| - | 848 | 19.5 | Not in COGs |
1 The total is based on the total number of protein coding genes in the genome.
Table 3.
Putative gene clusters coding for secondary metabolites in Bacillus sp. 1839.
| Type | From (bp) | To (bp) | Most Similar Known Cluster | % Similarity | Accession Number |
|---|---|---|---|---|---|
| Terpene | 348,477 | 367,415 | Cytobacillus gottheilii strain FJAT-2394 | 88 | NZ_KV440945 |
| Thiopeptide-Linear azol(in)e-containing peptides | 447,791 | 476,894 | 82 | ||
| Terpene | 973,108 | 1,001,908 | 89 | ||
| Type III polyketide synthase cluster | 2,777,409 | 2,818,491 | 100 |
Table 4.
General genome features of the Cytobacillus spp. closely related to Bacillus sp. 1839.
| Organism | NCBI Accession No. | Assembly Level | Size (bp) | GC (%) | Predicted Coding Sequences | No. of Genes | No. of Proteins | No. of RNAs |
|---|---|---|---|---|---|---|---|---|
| Cytobacillus gottheilii Marseille-P3555 | NZ_FUVC00000000.1 | Scaffold | 4,719,939 | 39 | 4492 | 4621 | 4452 | 129 |
| Cytobacillus gottheilii FJAT-2394 | NZ_KV440945 | Genome | 4,584,535 | 39 | 4370 | 4475 | 4310 | 105 |
Table 5.
Average nucleotide identity (ANI) and tetranucleotide frequency correlation coefficient (Tetra) analysis of Bacillus sp. 1839 and closely related Cytobacillus strains.
Table 5.
Average nucleotide identity (ANI) and tetranucleotide frequency correlation coefficient (Tetra) analysis of Bacillus sp. 1839 and closely related Cytobacillus strains.
| Bacillus sp. 1839 | Cytobacillus gottheilii Marseille-P3555 | Cytobacillus gottheilii FJAT-2394 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ANIb | Aligned | Tetra | ANIb | Aligned | Tetra | ANIb | Aligned | Tetra | |
| Bacillus sp. 1839 | * | 97.55 | 85.95 | 0.99891 | 97.55 | 85.95 | 0.99892 | ||
| Cytobacillusgottheilii Marseille-P3555 | 97.32 | 86.16 | 0.99891 | * | 100.00 | 99.49 | 1.0 | ||
| Cytobacillus gottheilii FJAT-2394 | 97.34 | 86.18 | 0.99892 | 100.00 | 99.47 | 1.0 | * | ||
* The asterisk indicates that the strain is compared to itself.
Table 6.
Digital DNA–DNA hybridization (dDDH) analysis of Bacillus sp. 1839 with closely related Cytobacillus strains.
Table 6.
Digital DNA–DNA hybridization (dDDH) analysis of Bacillus sp. 1839 with closely related Cytobacillus strains.
| Query Genome | Reference Genome | Formula 1 | Formula 2 | Formula 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DDH (%) | Model C.I. (%) | Distance | DDH (%) | Model C.I. (%) | Distance | DDH (%) | Model C.I. (%) | Distance | ||
| Bacillus sp. 1839 | Cytobacillus gottheilii Marseille-P3555 | 83.7 | 79.8–86.9 | 0.1140 | 79.6 | 76.6–82.2 | 0.0239 | 85.9 | 82.8–88.5 | 0.1351 |
| Bacillus sp. 1839 | Cytobacillus gottheilii JAT-2394 | 83.7 | 79.9–86.9 | 0.1137 | 79.6 | 76.6–82.2 | 0.0239 | 85.9 | 82.8–88.6 | 0.1349 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Melnikova, D.I.; Nijland, R.; Magarlamov, T.Y. The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium. Toxins 2021, 13, 410. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060410
AMA Style
Melnikova DI, Nijland R, Magarlamov TY. The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium. Toxins. 2021; 13(6):410. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060410
Chicago/Turabian StyleMelnikova, Daria I., Reindert Nijland, and Timur Yu. Magarlamov. 2021. "The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium" Toxins 13, no. 6: 410. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060410
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.