Tamoxifen Derivatives Alter Retromer-Dependent Endosomal Tubulation and Sorting to Block Retrograde Trafficking of Shiga Toxins

 ,
,
Abstract
:1. Introduction
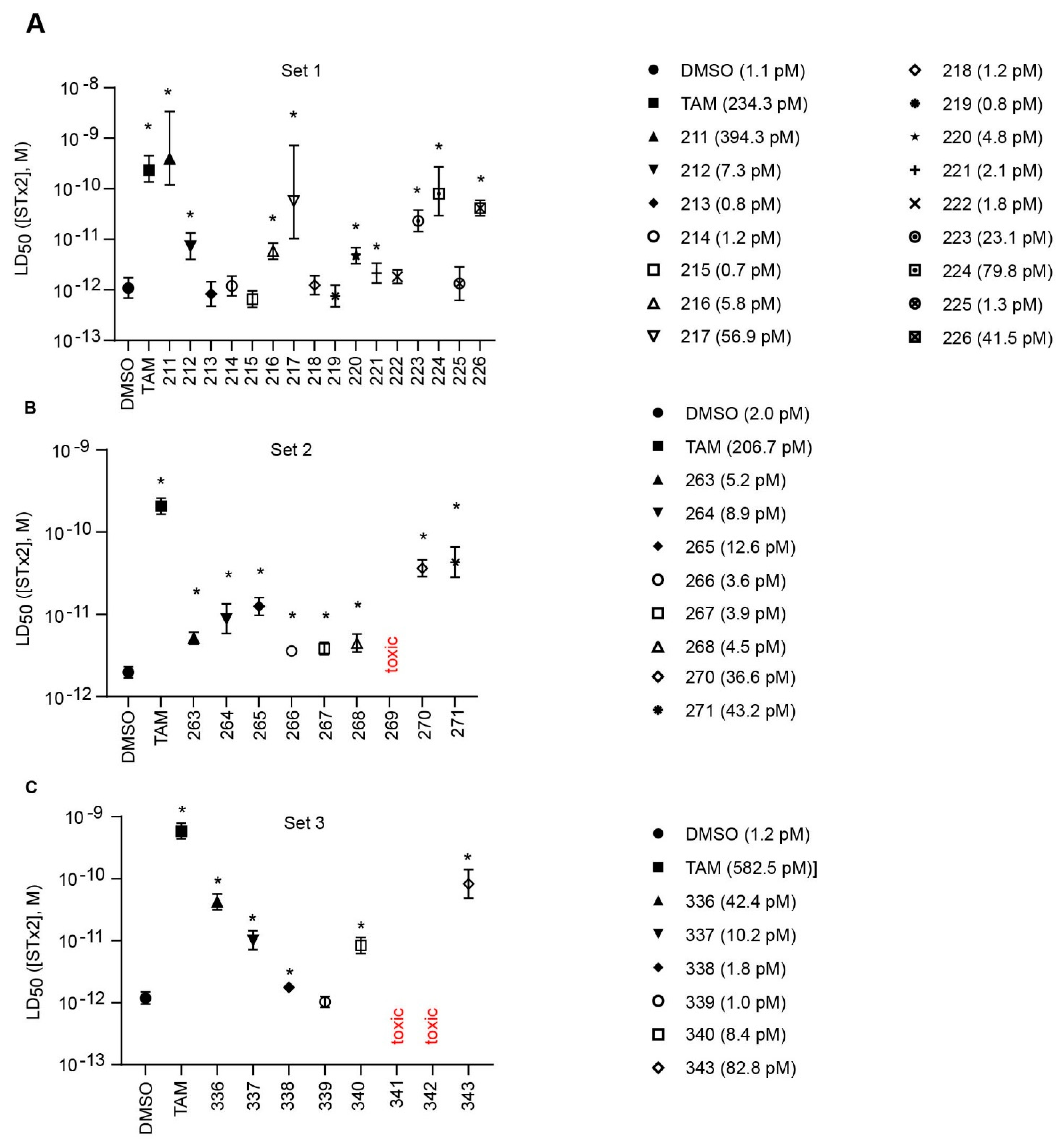
2. Results
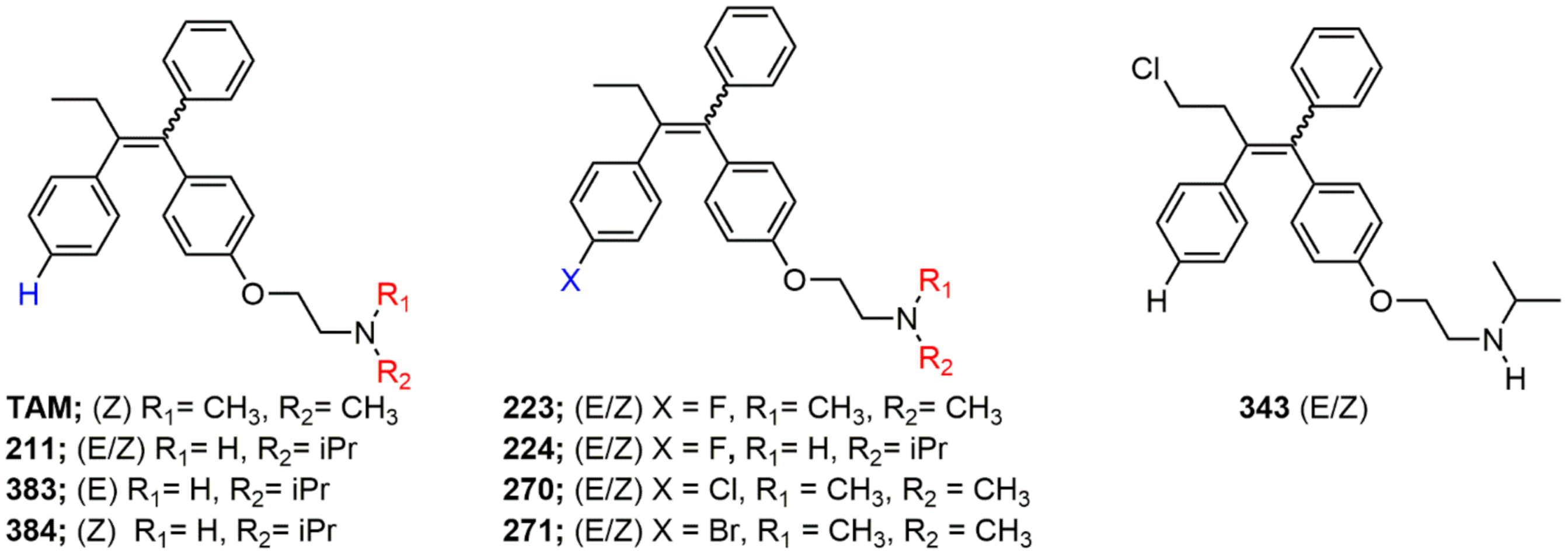
2.1. Structure Activity Relationships
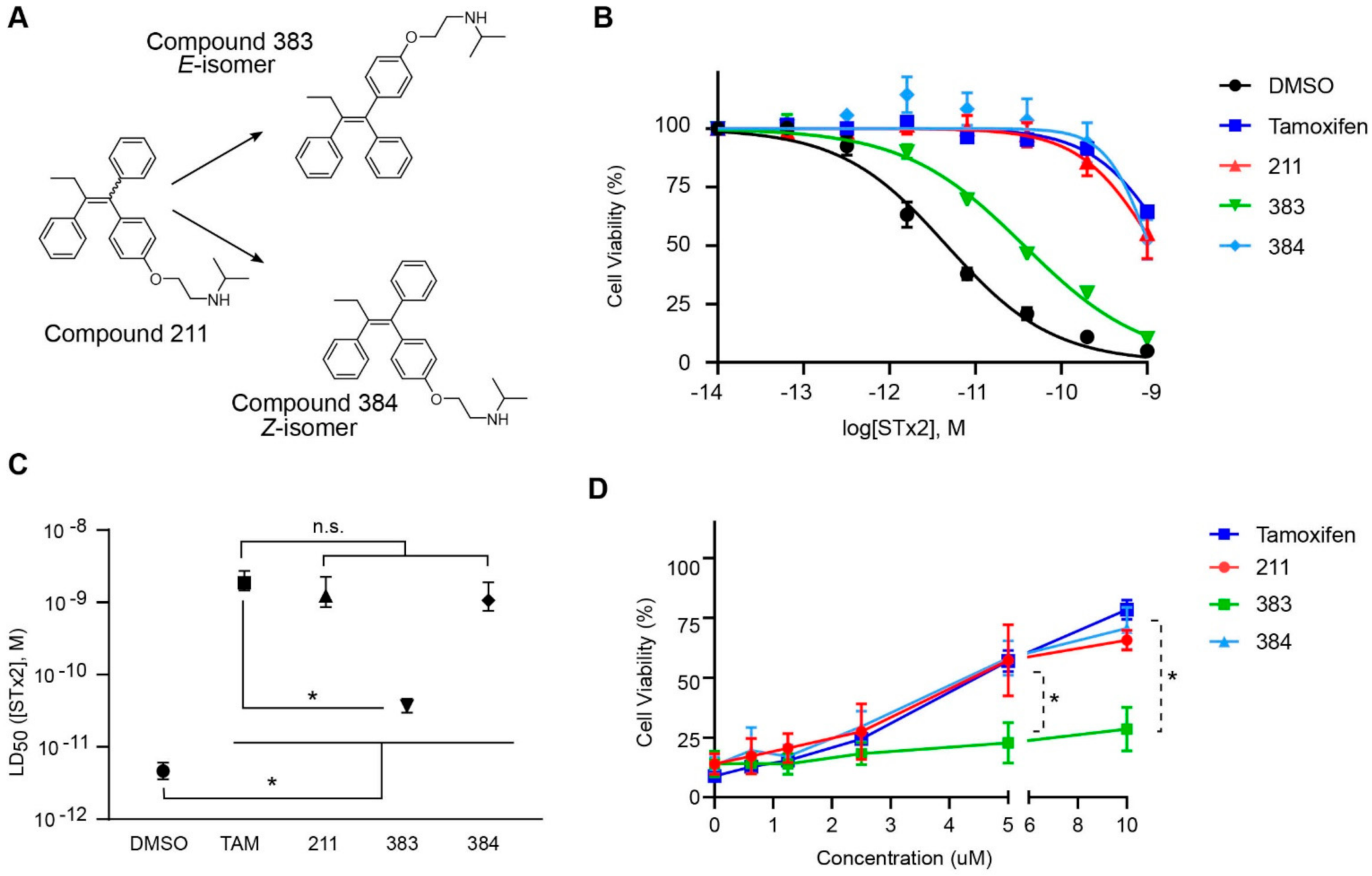
2.2. The Protective Function of Compound 211 Is Stereospecific
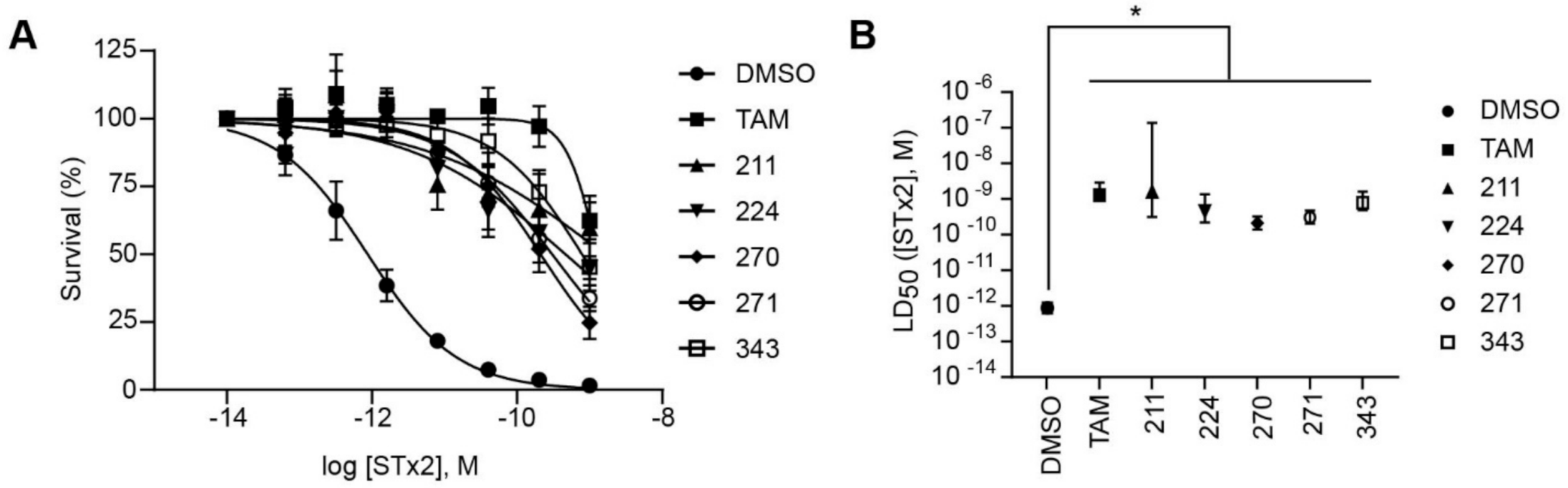
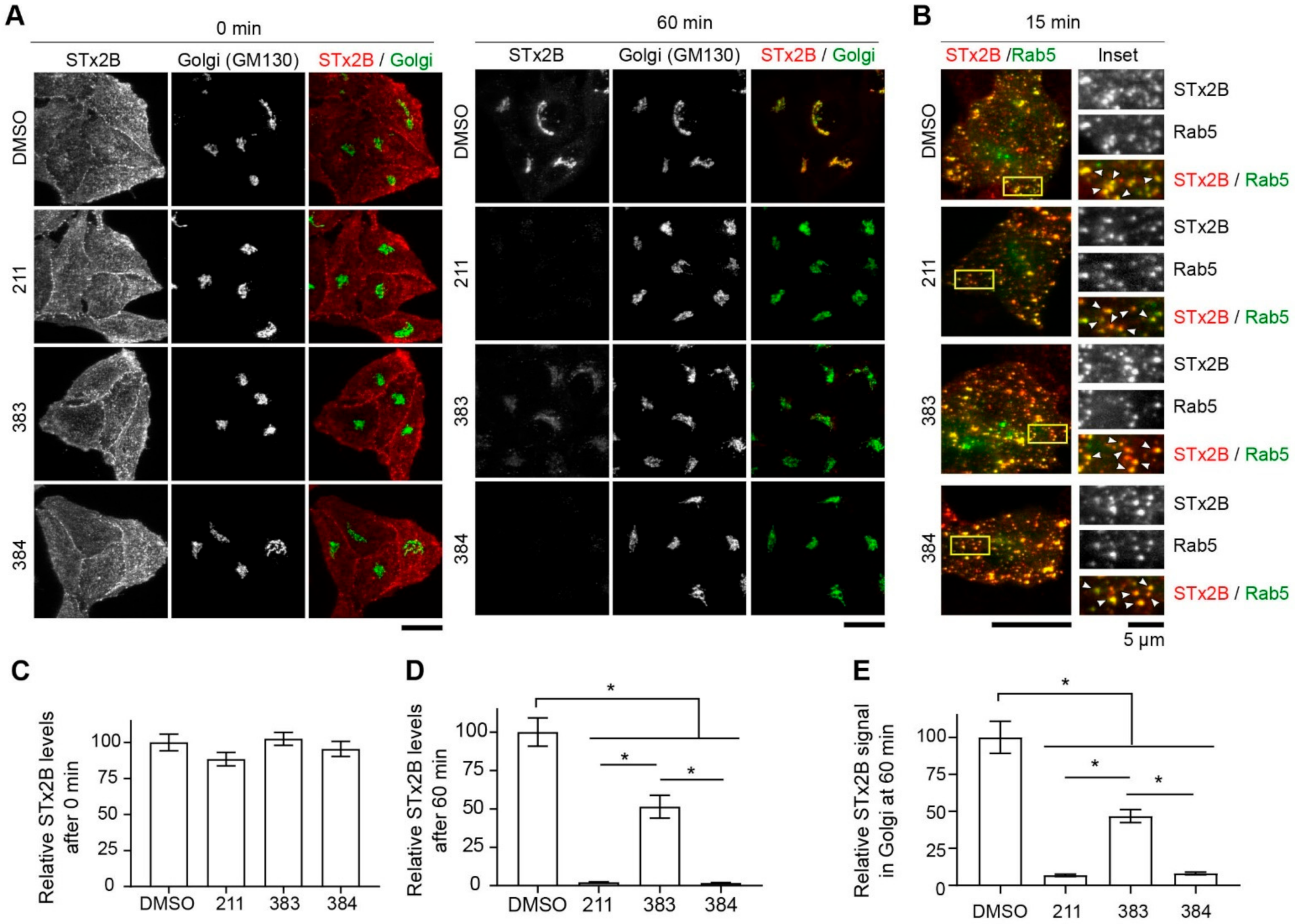
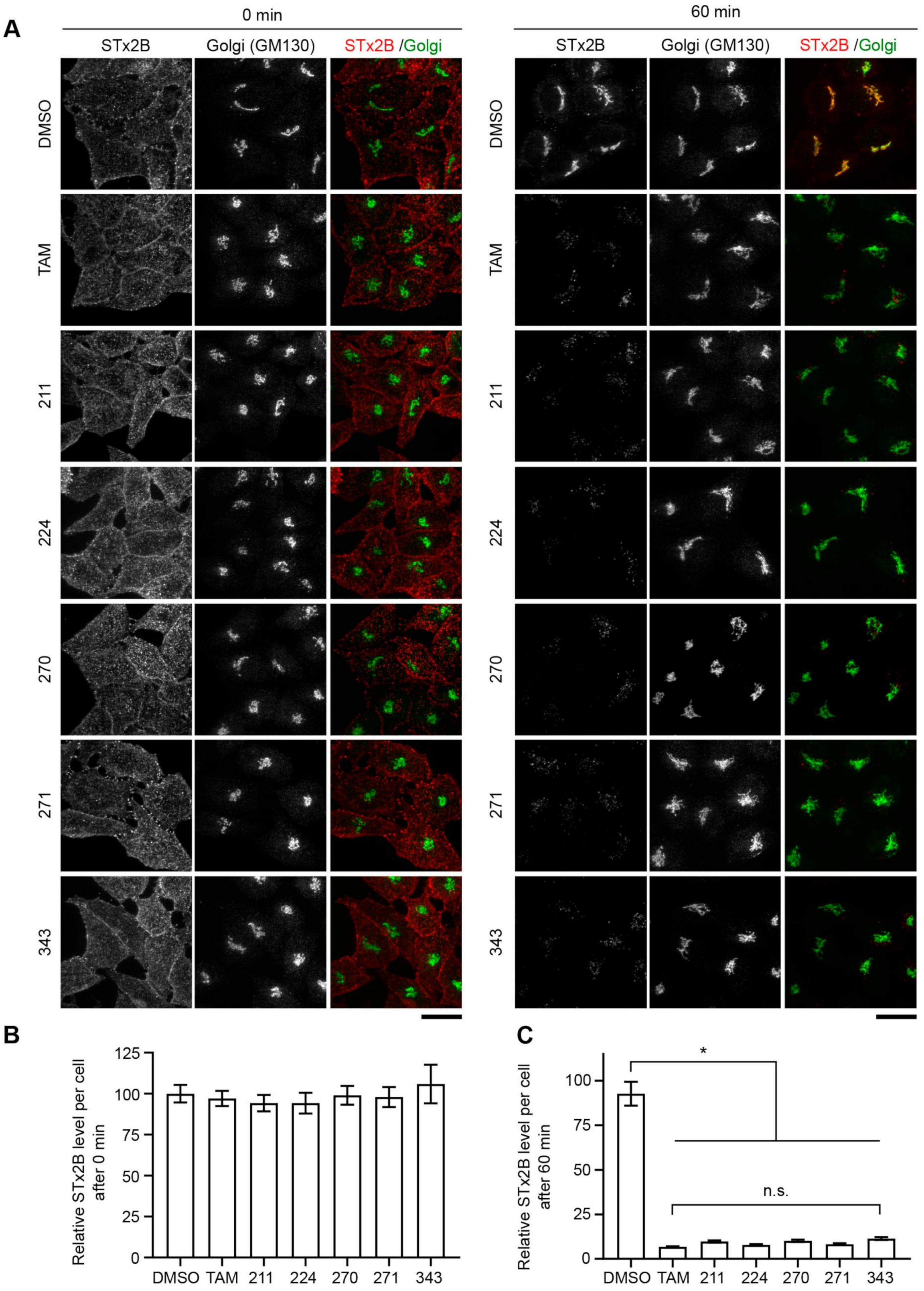
2.3. Protective Compounds Block Retrograde Trafficking of STx2
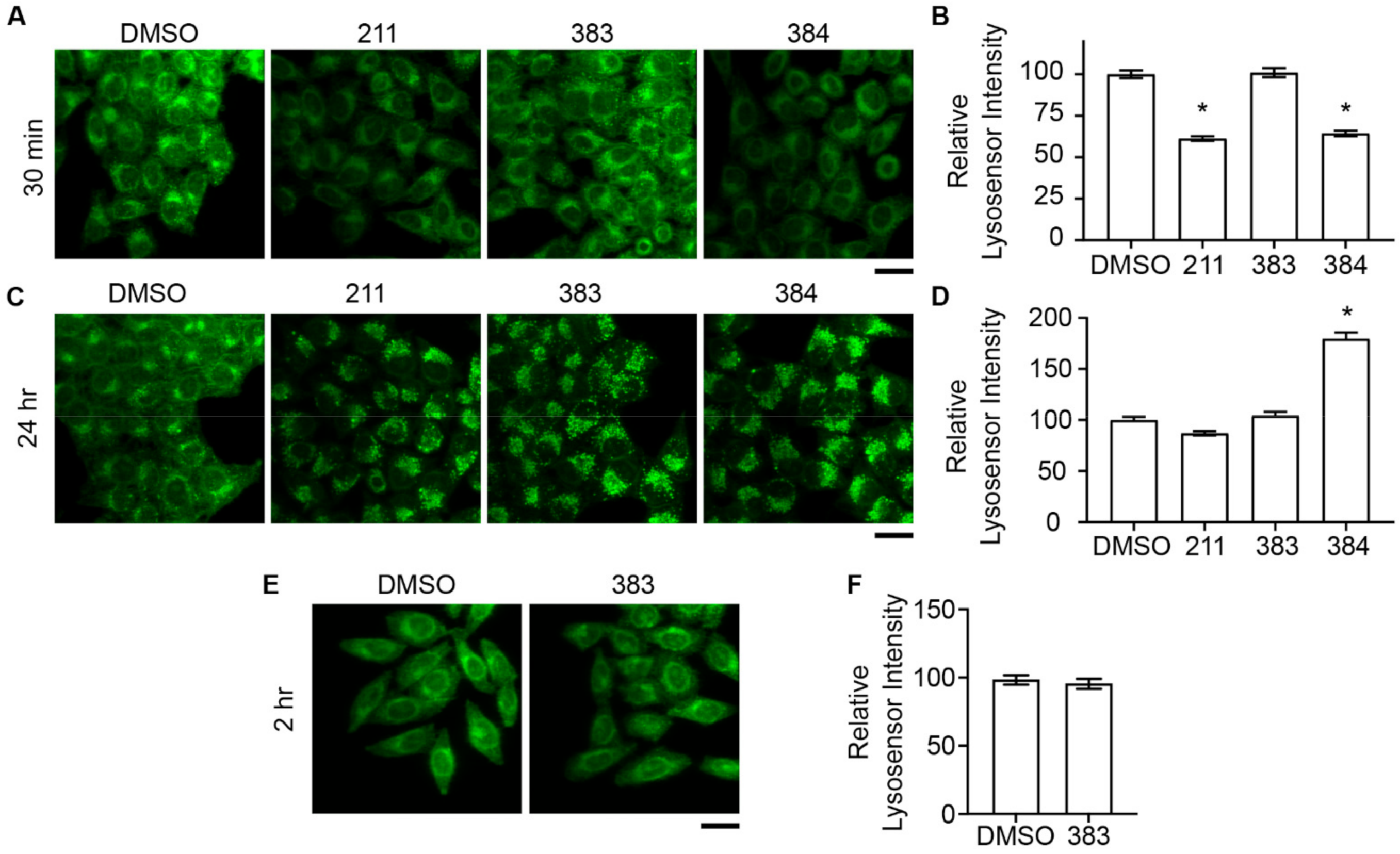
2.4. Relationship between Deacidification of Endolysosomes and Inhibition of STx2 Trafficking
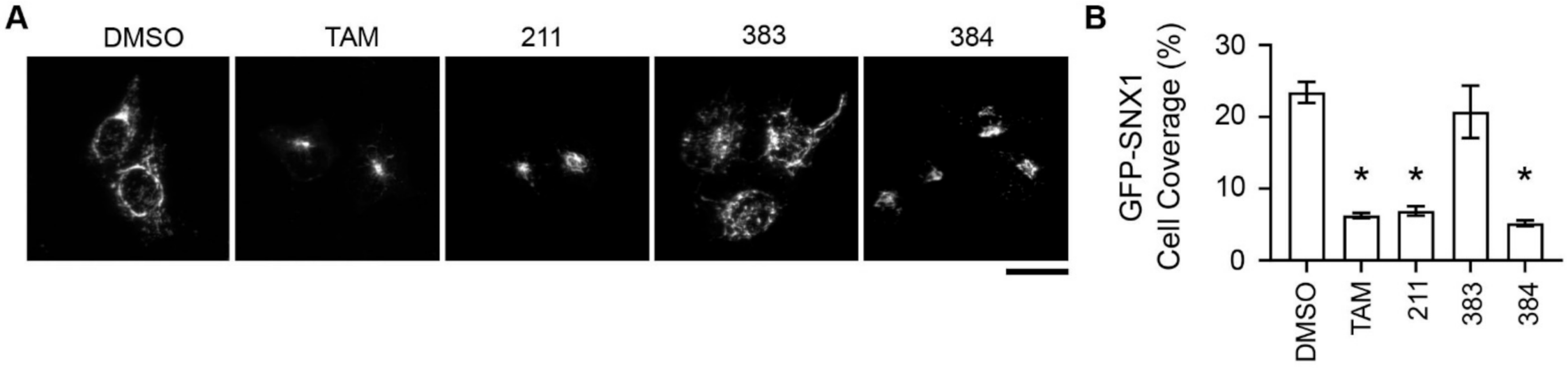
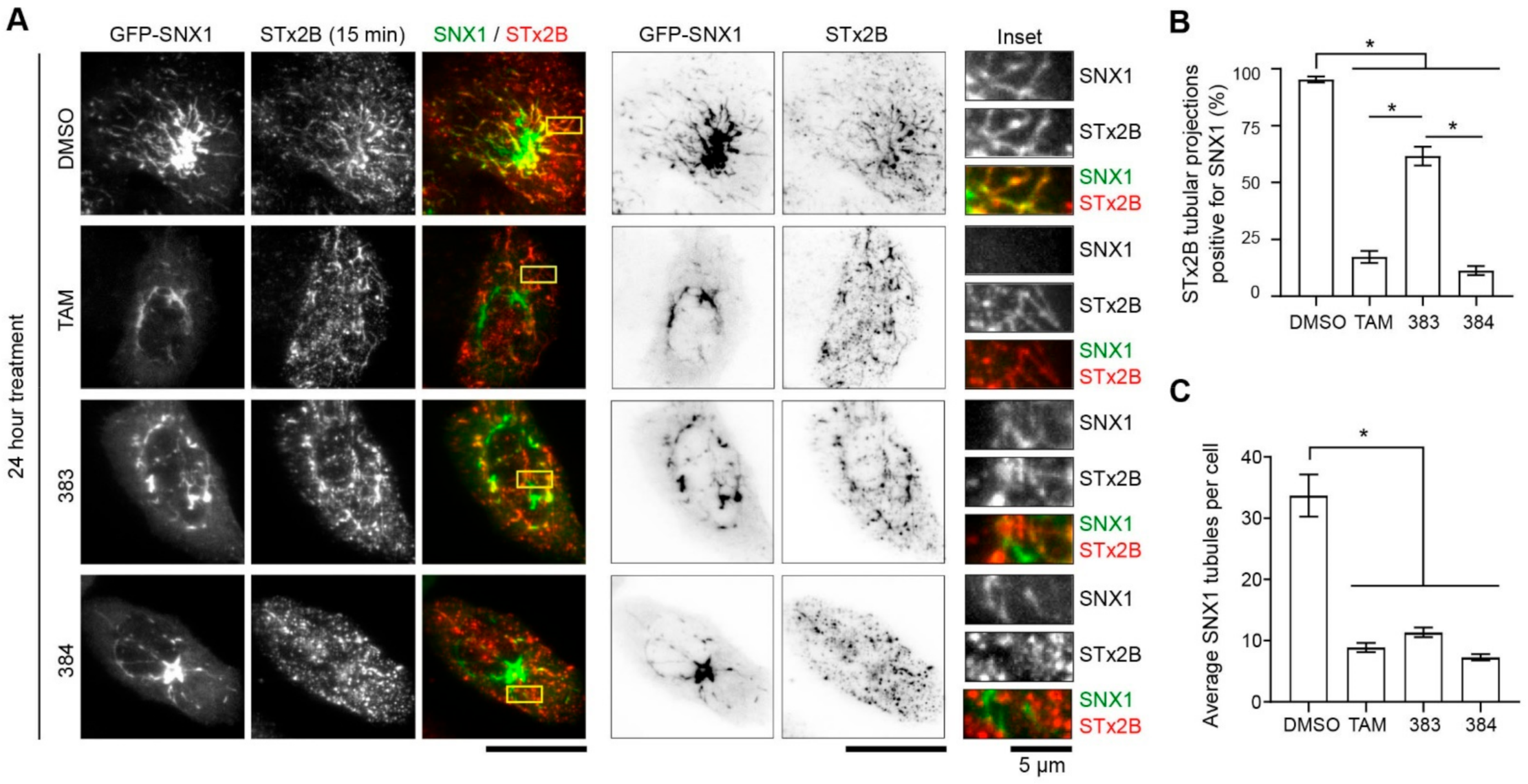
2.5. Tamoxifen Compounds Alter Retromer-Mediated Endosomal Tubulation and Sorting
3. Discussion
4. Materials and Methods
4.1. Generation of Novel Tamoxifen Derivatives
4.2. Plasmids and Reagents
4.3. Cell Culture and Transfections
4.4. Cytotoxicity Assay
4.5. STx2B Transport Assay
4.6. Immunofluorescence Microscopy, Imaging, Quantitative Analysis, and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-Related Illness and Death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Pickering, L.K. Oski’s Paediatrics: Principles and Practice, 3rd ed.; McMillan, J.A., Feigin, R.D., DeAngelis, C., Jones, M.D., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 1063–1068. [Google Scholar]
- Obrig, T.G. Escherichia coli Shiga Toxin Mechanisms of Action in Renal Disease. Toxins 2010, 2, 2769–2794. [Google Scholar] [CrossRef] [Green Version]
- Majowicz, S.E.; Scallan, E.; Jones-Bitton, A.; Sargeant, J.M.; Stapleton, J.; Angulo, F.J.; Yeung, D.H.; Kirk, M.D. Global Incidence of Human Shiga Toxin–Producing Escherichia coli Infections and Deaths: A Systematic Review and Knowledge Synthesis. Foodborne Pathog. Dis. 2014, 11, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between Virulence Factors of Shiga Toxin-Producing Escherichia coli and Disease in Humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushiro, A.; Sato, K.; Miyamoto, H.; Yamamura, T.; Honda, T. Induction of Prophages of Enterohemorrhagic Escherichia coli O157:H7 with Norfloxacin. J. Bacteriol. 1999, 181, 2257–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGannon, C.M.; Fuller, C.A.; Weiss, A.A. Different Classes of Antibiotics Differentially Influence Shiga Toxin Production. Antimicrob. Agents Chemother. 2010, 54, 3790–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walterspiel, J.N.; Morrow, A.L.; Cleary, T.G.; Ashkenazi, S. Effect of subinhibitory concentrations of antibiotics on extracellular shiga-like toxin I. Infection 1992, 20, 25–29. [Google Scholar] [CrossRef]
- Zhang, X.; McDaniel, A.D.; Wolf, L.E.; Keusch, G.T.; Waldor, M.K.; Acheson, D.W.K. Quinolone Antibiotics Induce Shiga Toxin–Encoding Bacteriophages, Toxin Production, and Death in Mice. J. Infect. Dis. 2000, 181, 664–670. [Google Scholar] [CrossRef]
- Jenko, K.L.; Zhang, Y.; Kostenko, Y.; Fan, Y.; Garcia-Rodriguez, C.; Lou, J.; Marks, J.D.; Varnum, S.M. Development of an ELISA microarray assay for the sensitive and simultaneous detection of ten biodefense toxins. Analyst 2014, 139, 5093–5102. [Google Scholar] [CrossRef]
- Beddoe, T.; Paton, A.W.; Le Nours, J.; Rossjohn, J.; Paton, J.C. Structure, biological functions and applications of the AB5 toxins. Trends Biochem. Sci. 2010, 35, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Linstedt, A.D. Retrograde trafficking of AB5 toxins: Mechanisms to therapeutics. J. Mol. Med. 2013, 91, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Selyunin, A.; Mukhopadhyay, S. Targeting the Early Endosome-to-Golgi Transport of Shiga Toxins as a Therapeutic Strategy. Toxins 2020, 12, 342. [Google Scholar] [CrossRef]
- Mallard, F.; Antony, C.; Tenza, D.; Salamero, J.; Goud, B.; Johannes, L. Direct Pathway from Early/Recycling Endosomes to the Golgi Apparatus Revealed through the Study of Shiga Toxin B-fragment Transport. J. Cell Biol. 1998, 143, 973–990. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Linstedt, A.D. Manganese Blocks Intracellular Trafficking of Shiga Toxin and Protects Against Shiga Toxicosis. Science 2012, 335, 332–335. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Redler, B.; Linstedt, A.D. Shiga toxin–binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Mol. Biol. Cell 2013, 24, 2311–2318. [Google Scholar] [CrossRef]
- Selyunin, A.S.; Mukhopadhyay, S. A Conserved Structural Motif Mediates Retrograde Trafficking of Shiga Toxin Types 1 and 2. Traffic 2015, 16, 1270–1287. [Google Scholar] [CrossRef] [Green Version]
- Saenz, J.B.; Doggett, T.A.; Haslam, D.B. Identification and Characterization of Small Molecules That Inhibit Intracellular Toxin Transport. Infect. Immun. 2007, 75, 4552–4561. [Google Scholar] [CrossRef] [Green Version]
- Stechmann, B.; Bai, S.-K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of Retrograde Transport Protects Mice from Lethal Ricin Challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef]
- Selyunin, A.S.; Hutchens, S.; McHardy, S.F.; Mukhopadhyay, S. Tamoxifen blocks retrograde trafficking of Shiga toxin 1 and 2 and protects against lethal toxicosis. Life Sci. Alliance 2019, 2, e201900439. [Google Scholar] [CrossRef]
- Selyunin, A.S.; Iles, L.R.; Bartholomeusz, G.; Mukhopadhyay, S. Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking. J. Cell Biol. 2017, 216, 3249–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Van Weert, A.W.; Dunn, K.W.; Gueze, H.J.; Maxfield, F.R.; Stoorvogel, W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J. Cell Biol. 1995, 130, 821–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzeroni, M.; Serrano, D.; Dunn, B.K.; Heckman-Stoddard, B.M.; Lee, O.; Khan, S.; Decensi, A. Oral low dose and topical tamoxifen for breast cancer prevention: Modern approaches for an old drug. Breast Cancer Res. 2012, 14, 1–11. [Google Scholar] [CrossRef]
- Robinson, S.P.; Langan-Fahey, S.M.; Johnson, D.A.; Jordan, V.C. Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab. Dispos. 1991, 19, 36–43. [Google Scholar]
- Morello, K.C.; Wurz, G.T.; DeGregorio, M.W. Pharmacokinetics of Selective Estrogen Receptor Modulators. Clin. Pharmacokinet. 2003, 42, 361–372. [Google Scholar] [CrossRef]
- Altan, N.; Chen, Y.; Schindler, M.; Simon, S.M. Tamoxifen inhibits acidification in cells independent of the estrogen receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 4432–4437. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Schindler, M.; Simon, S.M. A Mechanism for Tamoxifen-mediated Inhibition of Acidification. J. Biol. Chem. 1999, 274, 18364–18373. [Google Scholar] [CrossRef] [Green Version]
- Utskarpen, A.; Slagsvold, H.H.; Dyve, A.B.; Skånland, S.S.; Sandvig, K. SNX1 and SNX2 mediate retrograde transport of Shiga toxin. Biochem. Biophys. Res. Commun. 2007, 358, 566–570. [Google Scholar] [CrossRef]
- Bujny, M.V.; Popoff, V.; Johannes, L.; Cullen, P. The retromer component sorting nexin-1 is required for efficient retrograde transport of Shiga toxin from early endosome to the trans Golgi network. J. Cell Sci. 2007, 120, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Popoff, V.; Mardones, G.A.; Tenza, D.; Rojas, R.; Lamaze, C.; Bonifacino, J.S.; Raposo, G.; Johannes, L. The retromer complex and clathrin define an early endosomal retrograde exit site. J. Cell Sci. 2007, 120, 2022–2031. [Google Scholar] [CrossRef] [Green Version]
- Burd, C.; Cullen, P.J. Retromer: A Master Conductor of Endosome Sorting. Cold Spring Harb. Perspect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef]
- Rojas, R.; Van Vlijmen, T.; Mardones, G.A.; Prabhu, Y.; Rojas, A.L.; Mohammed, S.; Heck, A.; Raposo, G.; Van Der Sluijs, P.; Bonifacino, J.S. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 2008, 183, 513–526. [Google Scholar] [CrossRef] [Green Version]
- Van Weering, J.R.; Verkade, P.; Cullen, P. SNX-BAR-Mediated Endosome Tubulation is Co-ordinated with Endosome Maturation. Traffic 2011, 13, 94–107. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab Conversion as a Mechanism of Progression from Early to Late Endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Abdellatif, K.R.; Belal, A.; Omar, H. Design, synthesis and biological evaluation of novel triaryl (Z)-olefins as tamoxifen analogues. Bioorg. Med. Chem. Lett. 2013, 23, 4960–4963. [Google Scholar] [CrossRef]
- Kelly, P.M.; Keely, N.O.; Bright, S.A.; Yassin, B.; Ana, G.; Fayne, D.; Zisterer, D.M.; Meegan, M.J. Novel Selective Estrogen Receptor Ligand Conjugates Incorporating Endoxifen-Combretastatin and Cyclofenil-Combretastatin Hybrid Scaffolds: Synthesis and Biochemical Evaluation. Molecules 2017, 22, 1440. [Google Scholar] [CrossRef]
- Agouridas, V.; Laı, I.; Cleeren, A.; Kizilian, E.; Magnier, E.; Blazejewski, J.-C.; Leclercq, G. Loss of antagonistic activity of tamoxifen by replacement of one N-methyl of its side chain by fluorinated residues. Bioorganic Med. Chem. 2006, 14, 7531–7538. [Google Scholar] [CrossRef]
- Robertson, D.W.; Katzenellenbogen, J.A.; Hayes, J.R.; Katzenellenbogen, B.S. Antiestrogen basicity--activity relationships: A comparison of the estrogen receptor binding and antiuterotrophic potencies of several analogues of (Z)-1,2-diphenyl-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-butene (tamoxifen, Nolvadex) having altered basicity. J. Med. Chem. 1982, 25, 167–171. [Google Scholar]
- Robertson, D.W.; Katzenellenbogen, J.A. Synthesis of the E-Isomers and Z-Isomers of the Anti-Estrogen Tamoxifen and Its Metabolite, Hydroxytamoxifen, in Tritium-Labeled Form. J. Org. Chem. 1982, 47, 2387–2393. [Google Scholar] [CrossRef]
- Shiina, I.; Sano, Y.; Nakata, K.; Kikuchi, T.; Sasaki, A.; Ikekita, M.; Nagahara, Y.; Hasome, Y.; Yamori, T.; Yamazaki, K. Synthesis and pharmacological evaluation of the novel pseudo-symmetrical tamoxifen derivatives as anti-tumor agents. Biochem. Pharmacol. 2008, 75, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.M.P.J.; Quezada, E.; Fajin, J.L.C.; Cordeiro, M.N.D.S.; Garrido, E.M.P.J.; Borges, F. Electrochemical Oxidation of Tamoxifen Revisited. Int. J. Electrochem. Sci. 2013, 8, 5710–5723. [Google Scholar]
- Childers, W.; Fan, R.; Martinez, R.; Colussi, D.J.; Melenski, E.; Liu, Y.; Gordon, J.; Abou-Gharbia, M.; Jacobson, M.A. Novel compounds that reverse the disease phenotype in Type 2 Gaucher disease patient-derived cells. Bioorganic Med. Chem. Lett. 2020, 30, 126806. [Google Scholar] [CrossRef] [PubMed]
- Cahiez, G.; Moyeux, A.; Poizat, M. Stereoselective synthesis of triarylethylenes via copper–palladium catalyzed decarboxylative cross-coupling: Synthesis of (Z)-tamoxifen. Chem. Commun. 2014, 50, 8982–8984. [Google Scholar] [CrossRef]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [Green Version]
- Carlton, J.; Bujny, M.; Peter, B.J.; Oorschot, V.M.; Rutherford, A.; Mellor, H.; Klumperman, J.; McMahon, H.T.; Cullen, P. Sorting Nexin-1 Mediates Tubular Endosome-to-TGN Transport through Coincidence Sensing of High- Curvature Membranes and 3-Phosphoinositides. Curr. Biol. 2004, 14, 1791–1800. [Google Scholar] [CrossRef] [Green Version]
- Touitou, I.; Mathieu, M.; Rochefort, H. Stable transfection of the estrogen receptor cDNA into Hela cells induces estrogen responsiveness of endogenous cathepsin D gene but not of cell growth. Biochem. Biophys. Res. Commun. 1990, 169, 109–115. [Google Scholar] [CrossRef]
- Komi, J.; Heikkinen, J.; Rutanen, M.; Halonen, K.; Lammintausta, R.; Ylikorkala, O. Effects of ospemifene, a novel SERM, on biochemical markers of bone turnover in healthy postmenopausal women. Gynecol. Endocrinol. 2004, 18, 152–158. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule Name | Structure | Lot ID | Molecule Name | Structure | Lot ID |
|---|---|---|---|---|---|
| CIDD-0000574 1 |  | Prestw-146 905 466 SM2019-96-70b1 | CIDD-0150264 |  | SM2019-96-107b |
| CIDD-0150211 |  | SM2019-96-67b SM2019-96-150c SM2019-96-172c1 | CIDD-0150265 |  | SM2019-96-111b |
| CIDD-0150212 |  | SM2019-96-69b | CIDD-0150266 |  | SM2019-96-113b |
| CIDD-0150213 |  | SM2019-96-71a | CIDD-0150267 |  | SM2019-96-120b |
| CIDD-0150214 |  | SM2019-96-72b | CIDD-0150268 |  | SM2019-96-102-2b |
| CIDD-0150215 |  | SM2019-96-73b | CIDD-0150269 |  | SM2019-96-134b |
| CIDD-0150216 |  | SM2019-96-74b | CIDD-0150270 |  | SM2019-96-132b |
| CIDD-0150217 |  | SM2019-96-75b2 | CIDD-0150271 |  | SM2019-96-133b |
| CIDD-0150218 |  | SM2019-96-76b1 | CIDD-0150336 |  | SM2019-96-138b |
| CIDD-0150219 |  | SM2019-96-78a2 | CIDD-0150337 |  | SM2019-96-139b |
| CIDD-0150220 |  | SM2019-96-79a | CIDD-0150338 |  | SM2019-96-140b |
| CIDD-0150221 |  | SM2019-96-80b | CIDD-0150339 |  | SM2019-96-147b |
| CIDD-0150222 |  | SM2019-96-81a | CIDD-0150340 |  | SM2019-96-143b |
| CIDD-0150223 |  | SM2019-96-83c | CIDD-0150341 |  | SM2019-96-144b |
| CIDD-0150224 |  | SM2019-96-84b | CIDD-0150342 |  | SM2019-96-145b |
| CIDD-0150225 |  | SM2019-96-85b | CIDD-0150343 |  | SM2019-96-148b1 |
| CIDD-0150226 |  | SM2019-96-86b | CIDD-0150383 |  | SM2019-96-150c1 |
| CIDD-0150263 |  | SM2019-96-92 | CIDD-0150384 |  | SM2019-96-150c4 |
| Compound | LD50 (95% CI) (pM) | Fold of DMSO (Different?) | Fold of TAM (Different?) |
|---|---|---|---|
| Figure 2A | |||
| DMSO | 1.1 (0.7–1.7) | - | 0.005 (Y) |
| TAM | 234.3 (136.0–454.4) | 213 (Y) | - |
| 211 | 394.3 (120.0–3407.0) | 358 (Y) | 1.68 (N) |
| 212 | 7.3 (4.0–13.4) | 6.64 (Y) | 0.031 (Y) |
| 213 | 0.8 (0.5–1.5) | 0.73 (N) | 0.003 (Y) |
| 214 | 1.2 (0.8–1.9) | 1.09 (N) | 0.005 (Y) |
| 215 | 0.7 (0.4–1.0) | 0.64 (N) | 0.003 (Y) |
| 216 | 5.8 (4.0–8.5) | 5.27 (Y) | 0.025 (Y) |
| 217 | 56.9 (10.4–723.0) | 51.7 (Y) | 0.243 (N) |
| 218 | 1.2 (0.8–1.9) | 1.09 (N) | 0.005 (Y) |
| 219 | 0.8 (0.5–1.2) | 0.73 (N) | 0.003 (Y) |
| 220 | 4.8 (3.3–6.8) | 4.36 (Y) | 0.020 (Y) |
| 221 | 2.1 (1.4–3.4) | 1.91 (Y) | 0.009 (Y) |
| 222 | 1.8 (1.3–2.5) | 1.64 (N) | 0.008 (Y) |
| 223 | 23.1 (14.2–37.8) | 21.0 (Y) | 0.099 (Y) |
| 224 | 79.8 (29.4–272.7) | 72.5 (Y) | 0.341 (N) |
| 225 | 1.3 (0.6–2.9) | 1.18 (N) | 0.006 (Y) |
| 226 | 41.5 (29.3–59.2) | 37.7 (Y) | 0.177 (Y) |
| Figure 2B | |||
| DMSO | 2.0 (1.7–2.3) | - | 0.010 (Y) |
| TAM | 206.7 (165.3–259.4) | 103 (Y) | - |
| 263 | 5.2 (4.4–6.1) | 2.60 (Y) | 0.025 (Y) |
| 264 | 8.9 (5.9–13.6) | 4.45 (Y) | 0.043 (Y) |
| 265 | 12.6 (9.8–16.2) | 6.30 (Y) | 0.061 (Y) |
| 266 | 3.6 (3.2–4.1) | 1.80 (Y) | 0.017 (Y) |
| 267 | 3.9 (3.2–4.6) | 1.95 (Y) | 0.019 (Y) |
| 268 | 4.5 (3.5–5.8) | 2.25 (Y) | 0.022 (Y) |
| 269 | toxic | - | - |
| 270 | 36.6 (29.0–46.2) | 18.3 (Y) | 0.177 (Y) |
| 271 | 43.2 (28.4–66.0) | 21.6 (Y) | 0.209 (Y) |
| Figure 2C | |||
| DMSO | 1.2 (1.0–1.5) | - | 0.002 (Y) |
| TAM | 582.5 (440.7–791.5) | 485 (Y) | - |
| 336 | 42.4 (31.6–57.0) | 35.3 (Y) | 0.073 (Y) |
| 337 | 10.2 (7.1–14.5) | 8.50 (Y) | 0.018 (Y) |
| 338 | 1.8 (1.6–2.0) | 1.50 (Y) | 0.003 (Y) |
| 339 | 1.0 (0.8–1.3) | 0.83 (N) | 0.002 (Y) |
| 340 | 8.4 (6.3–11.4) | 7.00 (Y) | 0.014 (Y) |
| 341 | toxic | - | - |
| 342 | toxic | - | - |
| 343 | 82.8 (48.7–141.2) | 69.0 (Y) | 0.142 (Y) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selyunin, A.S.; Nieves-Merced, K.; Li, D.; McHardy, S.F.; Mukhopadhyay, S. Tamoxifen Derivatives Alter Retromer-Dependent Endosomal Tubulation and Sorting to Block Retrograde Trafficking of Shiga Toxins. Toxins 2021, 13, 424. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060424
Selyunin AS, Nieves-Merced K, Li D, McHardy SF, Mukhopadhyay S. Tamoxifen Derivatives Alter Retromer-Dependent Endosomal Tubulation and Sorting to Block Retrograde Trafficking of Shiga Toxins. Toxins. 2021; 13(6):424. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060424
Chicago/Turabian StyleSelyunin, Andrey S., Karinel Nieves-Merced, Danyang Li, Stanton F. McHardy, and Somshuvra Mukhopadhyay. 2021. "Tamoxifen Derivatives Alter Retromer-Dependent Endosomal Tubulation and Sorting to Block Retrograde Trafficking of Shiga Toxins" Toxins 13, no. 6: 424. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060424