Acute Kidney Injury and Organ Dysfunction: What Is the Role of Uremic Toxins?

 , ,
, ,
Abstract
:1. Introduction
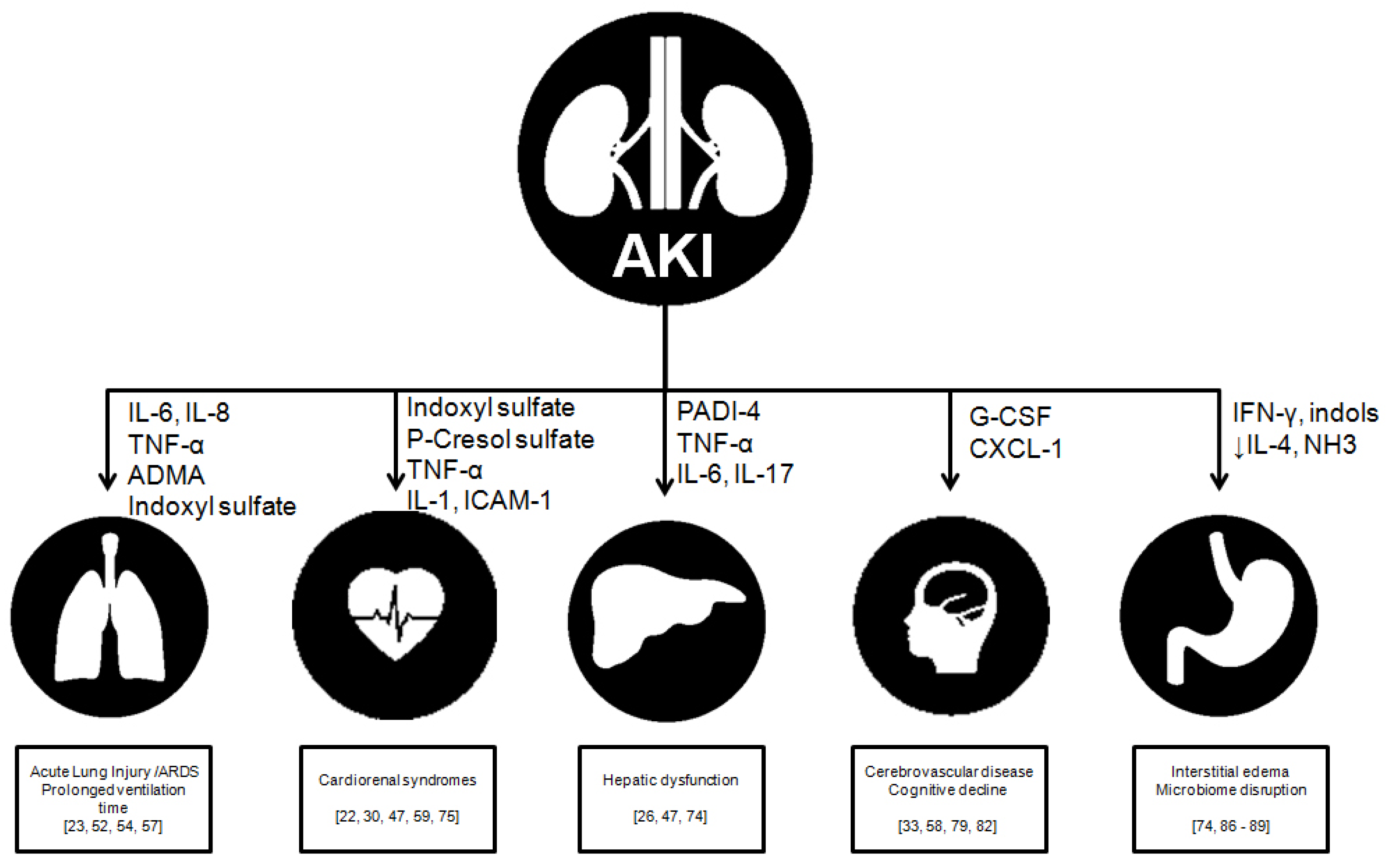
2. AKI: Beyond the Kidney
3. Small Water-Soluble Substances
3.1. Asymmetric Dimethyl-Arginine (ADMA)
3.2. Guanidine Compounds
3.3. Uric Acid
4. Protein-Bound Uremic Toxins
4.1. Indoxyl Sulfate (IS)
4.2. Homocysteine
5. Medium Molecular Weight Molecules
5.1. Proinflammatory Cytokines
5.2. Clinical Applications
6. Materials and Methods
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [Green Version]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Fitzgerald, D.B.R.L.; Mehta, D.C.E.M.R.; Endre, Z.; Forni, L.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.F.; Cohen, G.M.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelfarb, J.; McMonagle, E.; Freedman, S.; Klenzak, J.; McMenamin, E.; Ellen, M.; Pupim, L.B.; Ikizler, T. The PICARD Group Oxidative Stress Is Increased in Critically Ill Patients with Acute Renal Failure. J. Am. Soc. Nephrol. 2004, 15, 2449–2456. [Google Scholar] [CrossRef]
- Park, J.-S.; Kim, S.-J.; Lee, S.-W.; Lee, E.-J.; Han, K.-S.; Moon, S.-W.; Hong, Y.-S. Initial Low Oxygen Extraction Ratio Is Related to Severe Organ Dysfunction and High In-Hospital Mortality in Severe Sepsis and Septic Shock Patients. J. Emerg. Med. 2015, 49, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Kanagasundaram, N.S. Pathophysiology of ischaemic acute kidney injury. Ann. Clin. Biochem. Int. J. Lab. Med. 2015, 52, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okusa, M.D. The Changing Pattern of Acute Kidney Injury: From One to Multiple Organ Failure. Contrib. Nephrol. 2010, 165, 153–158. [Google Scholar] [CrossRef]
- Sutton, T.A.; Fisher, C.J.; Molitoris, B.A. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002, 62, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Van Laecke, S.; Glorieux, G. What is new in uremic toxicity? Pediatr. Nephrol. 2008, 23, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raff, A.C.; Meyer, T.W.; Hostetter, T.H. New insights into uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2008, 17, 560–565. [Google Scholar] [CrossRef]
- Lisowska-Myjak, B. Uremic Toxins and Their Effects on Multiple Organ Systems. Nephron 2014, 128, 303–311. [Google Scholar] [CrossRef]
- Sun, J.; Shannon, M.; Ando, Y.; Schnackenberg, L.K.; Khan, N.A.; Portilla, D.; Beger, R.D. Serum metabolomic profiles from patients with acute kidney injury: A pilot study. J. Chromatogr. B 2012, 893–894, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Popkov, V.A.; Silachev, D.N.; Zalevsky, A.O.; Zorov, D.B.; Plotnikov, E.Y. Mitochondria as a Source and a Target for Uremic Toxins. Int. J. Mol. Sci. 2019, 20, 3094. [Google Scholar] [CrossRef] [Green Version]
- Herget-Rosenthal, S.; Glorieux, G.; Jankowski, J.; Jankowski, V. Uremic Toxins in Acute Kidney Injury. Semin. Dial. 2009, 22, 445–448. [Google Scholar] [CrossRef]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am. J. Kidney Dis. 2018, 72, 846–856. [Google Scholar] [CrossRef]
- White, L.E.; Hassoun, H.T. Inflammatory Mechanisms of Organ Crosstalk during Ischemic Acute Kidney Injury. Int. J. Nephrol. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hassoun, H.T.; Santora, R.; Rabb, H. Organ crosstalk: The role of the kidney. Curr. Opin. Crit. Care 2009, 15, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T. ADMA metabolism and clearance. Vasc. Med. 2005, 10, S73–S81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhovershin, R.A.; Gilinskiĭ, M.A. The role of asymmetric dimethylarginine in the regulation of nitric oxide level in rats with acute renal injury. Ross. Fiziol. Zhurnal Im. Sechenova 2012, 98, 497–505. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular Biology of the Asymmetric Dimethylarginine:Dimethylarginine Dimethylaminohydrolase Pathway. Arter. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef]
- Ma, T.; Liu, X.; Liu, Z. Role of asymmetric dimethylarginine in rat acute lung injury induced by acute ischemic kidney injury. Mol. Med. Rep. 2015, 12, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Carnegie, P.; Fellows, F.; Symington, G. Urinary excretion of methylarginine in human disease. Metabolism 1977, 26, 531–537. [Google Scholar] [CrossRef]
- Nijveldt, R. Asymmetrical dimethylarginine (ADMA) in critically ill patients: High plasma ADMA concentration is an independent risk factor of ICU mortality. Clin. Nutr. 2003, 22, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijveldt, R.J.; Siroen, M.P.C.; Teerlink, T.; Van Leeuwen, P.A.M. Elimination of asymmetric dimethylarginine by the kidney and the liver: A link to the development of multiple organ failure? J. Nutr. 2004, 134, 2848S–2852S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wołyniec, W.; Kasprowicz, K.; Giebułtowicz, J.; Korytowska, N.; Zorena, K.; Bartoszewicz, M.; Rita-Tkachenko, P.; Renke, M.; Ratkowski, W.; Tkachenko, R. Changes in Water Soluble Uremic Toxins and Urinary Acute Kidney Injury Biomarkers After 10- and 100-km Runs. Int. J. Environ. Res. Public Health 2019, 16, 4153. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, D.R.; Meert, K.; Sarnaik, A.; Marescau, B.; Dedeyn, P.P. Guanidino Compound Metabolism in Arginine-Free Diet Induced Hyperammonemia. Enzyme 1991, 45, 128–136. [Google Scholar] [CrossRef]
- Levillain, O.; Parvy, P.; Hassler, C. Amino acid handling in uremic rats: Citrulline, a reliable marker of renal insufficiency and proximal tubular dysfunction. Metabolism 1997, 46, 611–618. [Google Scholar] [CrossRef]
- Glorieux, G.L.; Dhondt, A.W.; Jacobs, P.; Van Langeraert, J.; Lameire, N.H.; De Deyn, P.P.; Vanholder, R.C. In vitro study of the potential role of guanidines in leukocyte functions related to atherogenesis and infection. Kidney Int. 2004, 65, 2184–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R. Les toxines urémiques. Nephrologie 2003, 24, 373–376. [Google Scholar]
- Levillain, O.; Marescau, B.; Possemiers, I.; Banchaabouchi, M.; De Deyn, P. Influence of 72% injury in one kidney on several organs involved in guanidino compound metabolism: A time course study. Pflügers Arch. Eur. J. Physiol. 2001, 442, 558–569. [Google Scholar] [CrossRef]
- Shah, B.; Jagtap, P.; Sarmah, D.; Datta, A.; Raut, S.; Sarkar, A.; Bohra, M.; Singh, U.; Baidya, F.; Kalia, K.; et al. Cerebro-renal interaction and stroke. Eur. J. Neurosci. 2021, 53, 1279–1299. [Google Scholar] [CrossRef]
- Ejaz, A.A.; Mu, W.; Kang, D.-H.; Roncal, C.; Sautin, Y.Y.; Henderson, G.; Tabah-Fisch, I.; Keller, B.; Beaver, T.M.; Nakagawa, T.; et al. Could Uric Acid Have a Role in Acute Renal Failure? Clin. J. Am. Soc. Nephrol. 2006, 2, 16–21. [Google Scholar] [CrossRef]
- Mazzali, M.; Hughes, J.; Kim, Y.-G.; Jefferson, J.A.; Kang, D.-H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R. Elevated Uric Acid Increases Blood Pressure in the Rat by a Novel Crystal-Independent Mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef] [Green Version]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, C.E.M. Renal Effects of Hyperuricemia. Contrib. Nephrol. 2018, 192, 8–16. [Google Scholar] [CrossRef]
- Mercuro, G.; Vitale, C.; Cerquetani, E.; Zoncu, S.; Deidda, M.; Fini, M.; Rosano, G.M. Effect of hyperuricemia upon endothelial function in patients at increased cardiovascular risk. Am. J. Cardiol. 2004, 94, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Doehner, W.; Schoene, N.; Rauchhaus, M.; Leyva-Leon, F.; Pavitt, D.V.; Reaveley, D.A.; Schuler, G.; Coats, A.S.; Anker, S.D.; Hambrecht, R. Effects of Xanthine Oxidase Inhibition with Allopurinol on Endothelial Function and Peripheral Blood Flow in Hyperuricemic Patients with Chronic Heart Failure. Circulation 2002, 105, 2619–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, R.; Morris, A.D.; Belch, J.J.; Hill, A.; Struthers, A. Allopurinol Normalizes Endothelial Dysfunction in Type 2 Diabetics with Mild Hypertension. Hypertension 2000, 35, 746–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardillo, C.; Kilcoyne, C.M.; Cannon, R.O.; Quyyumi, A.A.; Panza, J.A. Xanthine Oxidase Inhibition with Oxypurinol Improves Endothelial Vasodilator Function in Hypercholesterolemic but Not in Hypertensive Patients. Hypertension 1997, 30, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-H.; Park, S.-K.; Lee, I.-K.; Johnson, R. Uric Acid–Induced C-Reactive Protein Expression: Implication on Cell Proliferation and Nitric Oxide Production of Human Vascular Cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, T.H.; Oppenheimer, S. Factors affecting urate excretion following diuretic administration in man. Am. J. Med. 1969, 47, 564–574. [Google Scholar] [CrossRef]
- Srivastava, A.; Palsson, R.; Leaf, D.; Higuera, A.; Chen, M.E.; Palacios, P.; Baron, R.M.; Sabbisetti, V.; Hoofnagle, A.N.; Vaingankar, S.M.; et al. Uric Acid and Acute Kidney Injury in the Critically Ill. Kidney Med. 2019, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S.; Kompa, A.; Krum, H. Protein-bound uremic toxins: A long overlooked culprit in cardiorenal syndrome. Am. J. Physiol. Physiol. 2016, 311, F52–F62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The Uremic Toxicity of Indoxyl Sulfate and p-Cresyl Sulfate: A Systematic Review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Lowenstein, J.; Nigam, S.K. Uremic Toxins in Organ Crosstalk. Front. Med. 2021, 8, 592602. [Google Scholar] [CrossRef]
- Wang, W.; Hao, G.; Pan, Y.; Ma, S.; Yang, T.; Shi, P.; Zhu, Q.; Xie, Y.; Ma, S.; Zhang, Q.; et al. Serum indoxyl sulfate is associated with mortality in hospital-acquired acute kidney injury: A prospective cohort study. BMC Nephrol. 2019, 20, 57. [Google Scholar] [CrossRef]
- Menez, S.; Hanouneh, M.; Shafi, T.; Jaar, B.G. Indoxyl sulfate is associated with mortality after AKI—More evidence needed! BMC Nephrol. 2019, 20, 280. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Ise, M.; Hirata, M.; Endo, K.; Ito, Y.; Seo, H.; Niwa, T. Indoxyl sulfate stimulates renal synthesis of transforming growth factor-beta 1 and progression of renal failure. Kidney Int. Suppl. 1997, 63, 211. [Google Scholar]
- Rabb, H.; Wang, Z.; Nemoto, T.; Hotchkiss, J.; Yokota, N.; Soleimani, M. Acute renal failure leads to dysregulation of lung salt and water channels. Kidney Int. 2003, 63, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumoto, M.; Kamobayashi, H.; Misato, Y.; Komori, M.; Yoshimura, M.; Hamada, A.; Kohda, Y.; Tomita, K.; Saito, H. Alleviation of cisplatin-induced acute kidney injury using phytochemical polyphenols is accompanied by reduced accumulation of indoxyl sulfate in rats. Clin. Exp. Nephrol. 2011, 15, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, N.; Sagata, M.; Saigo, C.; Yoneda, G.; Yamamoto, Y.; Nomura, Y.; Nishi, K.; Fujino, R.; Jono, H.; Saito, H. Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury. Int. J. Mol. Sci. 2016, 18, 11. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Shibahara, N.; Takagi, S.; Inoue, T.; Katsuoka, Y. AST-120 Treatment in Pre-Dialysis Period Affects the Prognosis in Patients on Hemodialysis. Ren. Fail. 2008, 30, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Yazawa, T.; Maeda, K.; Ise, M.; Sugano, M.; Kodama, T.; Uehara, Y. Effect of oral sorbent, AST-120, on serum con-centration of indoxyl sulfate in uremic rats. Nihon Jinzo Gakkai Shi 1990, 32, 695–701. [Google Scholar]
- Kreda, S.M.; Gynn, M.C.; Fenstermacher, D.A.; Boucher, R.C.; Gabriel, S.E. Expression and Localization of Epithelial Aquaporins in the Adult Human Lung. Am. J. Respir. Cell Mol. Biol. 2001, 24, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, K.; Watanabe, H.; Morisaki, T.; Matsuzaki, T.; Ohmura, T.; Hamada, A.; Saito, H. Involvement of Indoxyl Sulfate in Renal and Central Nervous System Toxicities During Cisplatin-induced Acute Renal Failure. Pharm. Res. 2007, 24, 662–671. [Google Scholar] [CrossRef]
- Fujii, H.; Nishijima, F.; Goto, S.; Sugano, M.; Yamato, H.; Kitazawa, R.; Kitazawa, S.; Fukagawa, M. Oral charcoal adsorbent (AST-120) prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol. Dial. Transplant. 2009, 24, 2089–2095. [Google Scholar] [CrossRef] [Green Version]
- Wu, V.-C.; The NSARF Group; Young, G.-H.; Huang, P.-H.; Lo, S.-C.; Wang, K.; Sun, C.-Y.; Liang, C.-J.; Huang, T.-M.; Chen, J.-H.; et al. In acute kidney injury, indoxyl sulfate impairs human endothelial progenitor cells: Modulation by statin. Angiogenesis 2013, 16, 609–624. [Google Scholar] [CrossRef]
- Veldeman, L.; Vanmassenhove, J.; Van Biesen, W.; Massy, Z.A.; Liabeuf, S.; Glorieux, G.; Vanholder, R. Evolution of protein-bound uremic toxins indoxyl sulphate and p-cresyl sulphate in acute kidney injury. Int. Urol. Nephrol. 2019, 51, 293–302. [Google Scholar] [CrossRef]
- Van Guldener, C.; Stam, F.; Stehouwer, C.D. Homocysteine metabolism in renal failure. Kidney Int. 2001, 59, 234–237. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Zhen, X.; Zhu, F.; Hu, Z.; Lei, W.; Li, S.; Zha, Y.; Nie, J. Hyperhomocysteinemia Exacerbates Cisplatin-induced Acute Kidney Injury. Int. J. Biol. Sci. 2017, 13, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Berger, K.; Moeller, M.J. Mechanisms of Epithelial Repair and Regeneration After Acute Kidney Injury. Semin. Nephrol. 2014, 34, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.; Hewitson, T.; Becker, G.J.; Jones, C.L. Ischemic acute renal failure: Long-term histology of cell and matrix changes in the rat. Kidney Int. 2000, 57, 2375–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-beta 1, TIMP-1 and pro-alpha 1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, 15. [Google Scholar]
- Miyazaki, T.; Aoyama, I.; Ise, M.; Seo, H.; Niwa, T. An oral sorbent reduces overload of indoxyl sulphate and gene expression of TGF-β1 in uraemic rat kidneys. Nephrol. Dial. Transplant. 2000, 15, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Tomosugi, N.I.; Naito, T.; Yokoyama, H.; Kobayashi, K.; Harada, A.; Mukaida, N.; Matsushima, K. Prevention of proteinuria by the administration of anti-interleukin 8 antibody in experimental acute immune complex-induced glomerulonephritis. J. Exp. Med. 1994, 180, 1135–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, A.P.; Mullin, J.M.; Knudsen, K.A.; Marano, C.W. Tissue remodeling during tumor necrosis factor-induced apoptosis in LLC-PK1 renal epithelial cells. Am. J. Physiol. Physiol. 1996, 270, F869–F879. [Google Scholar] [CrossRef]
- Shen, W.-C.; Liang, C.-J.; Huang, T.-M.; Liu, C.-W.; Wang, S.-H.; Young, G.-H.; Tsai, J.-S.; Tseng, Y.-C.; Peng, Y.-S.; Wu, V.-C.; et al. Indoxyl sulfate enhances IL-1β-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFκB/AP-1 pathway. Arch. Toxicol. 2015, 90, 2779–2792. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Hsu, Y.-H. IL-20 in Acute Kidney Injury: Role in Pathogenesis and Potential as a Therapeutic Target. Int. J. Mol. Sci. 2020, 21, 1009. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Chen, S.W.; Kim, M.; Brown, K.M.; Kolls, J.K.; D’Agati, V.D.; Lee, H.T. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab. Investig. 2010, 91, 63–84. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Hörl, W.H. Resistin as a Cardiovascular and Atherosclerotic Risk Factor and Uremic Toxin. Semin. Dial. 2009, 22, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Miller, L.; Ruiz-Velasco, V.; Kellum, J.A. Reversal of Acute Kidney Injury–Induced Neutrophil Dysfunction: A Critical Role for Resistin. Crit. Care Med. 2016, 44, e492–e501. [Google Scholar] [CrossRef] [Green Version]
- Shiao, C.-C.; Shiao, C.C.; Wu, P.C.; Huang, T.M.; Lai, T.S.; Yang, W.S.; Wu, C.H. Long-term remote organ consequences following acute kidney injury. Crit Car 2015, 19, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, C.; Bennis., Y.; Titeca-Beauport, D.; Caillard, P.; Cluet, Y.; Kamel, S. Two rapid, accurate liquid chromatography tandem mass spectrometry methods for the quantification of seven uremic toxins: An application for describing their ac-cumulation kinetic profile in a context of acute kidney injury. J. Chromatogr. B 2020, 1152, 122234. [Google Scholar] [CrossRef]
- Tanaka, S.; Okusa, M.D. Crosstalk between the nervous system and the kidney. Kidney Int. 2020, 97, 466–476. [Google Scholar] [CrossRef]
- Liu, M.; Liang, Y.; Chigurupati, S.; Lathia, J.D.; Pletnikov, M.; Sun, Z.; Crow, M.; Ross, C.A.; Mattson, M.P.; Rabb, H. Acute Kidney Injury Leads to Inflammation and Functional Changes in the Brain. J. Am. Soc. Nephrol. 2008, 19, 1360–1370. [Google Scholar] [CrossRef] [Green Version]
- Mair, R.D.; Nguyen, H.; Huang, T.-T.; Plummer, N.S.; Sirich, T.L.; Meyer, T.W. Accumulation of uremic solutes in the cerebrospinal fluid in experimental acute renal failure. Am. J. Physiol. Physiol. 2019, 317, F296–F302. [Google Scholar] [CrossRef]
- Tsai, H.-H.; Yen, R.-F.; Lin, C.-L.; Kao, C.-H. Increased risk of dementia in patients hospitalized with acute kidney injury: A nationwide population-based cohort study. PLoS ONE 2017, 12, e0171671. [Google Scholar] [CrossRef] [Green Version]
- Liu, S. Heart-kidney interactions: Mechanistic insights from animal models. Am. J. Physiol. Physiol. 2019, 316, F974–F985. [Google Scholar] [CrossRef]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate—Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Moreno, R.; Vincent, J.-L.; Matos, R.T.; Mendonça, A.; Cantraine, F.; Thijs, L.; Takala, J.; Sprung, C.; Antonelli, M.; Bruining, H.; et al. The use of maximum SOFA score to quantify organ dysfunction/failure in intensive care. Results of a prospective, multicentre study. Intensive Care Med. 1999, 25, 686–696. [Google Scholar] [CrossRef]
- Gong, J.; Noel, S.; Pluznick, J.L.; Hamad, A.R.A.; Rabb, H. Gut Microbiota-Kidney Cross-Talk in Acute Kidney Injury. Semin. Nephrol. 2019, 39, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Park, D.; Kim, Y.J.; Lee, I.; Kim, S.; Oh, C.; Kim, J.; Yang, J.; Jo, S. Lactobacillus salivarius BP121 prevents cisplatin-induced acute kidney injury by inhibition of uremic toxins such as indoxyl sulfate and p-cresol sulfate via alleviating dysbiosis. Int. J. Mol. Med. 2020, 45, 1130–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydzewska-Rosołowska, A.; Sroka, N.; Kakareko, K.; Rosołowski, M.; Zbroch, E.; Hryszko, T. The Links between Microbiome and Uremic Toxins in Acute Kidney Injury: Beyond Gut Feeling—A Systematic Review. Toxins 2020, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Chen, D.-Q.; Chen, L.; Liu, J.-R.; Vaziri, N.D.; Guo, Y.; Zhao, Y.-Y. Microbiome–metabolome reveals the contribution of gut–kidney axis on kidney disease. J. Transl. Med. 2019, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Small Water-Soluble Compounds (<500 D) with No KnownProtein Binding | Protein-Bound Compounds | Middle Molecules (>500 D) | ||
| ADMA1 1-Methyladenosine Guanidine Orotidine 1-Methylguanosine Guanidinoacetate Oxalate 1-Methylinosine Guanidinosuccinate Phenylacetylglutamine 8-OH-2′Deoxyguanosine Guanilin Phenylethylamine Hypoxanthine Pseudouridine α-keto-δ-Guanidinovaleriate Inosine SDMA α-N-Acetylarginine Malondialdehyde Sorbitol Arabinitol Mannitol Taurocyamine Argininic acid Methylguanidine Thiocyanate Benzylalcohol | Myoinositol Threitol β-Guanidinopropionate N2-Dimethylguanosine Trimethylamine Creatine N4-Acetylcytidine Thymine Creatinine N6-Methyladenosine Uracil Cytidine N6-Threonylcarbamoyladenosine Urea Dimethylglycine N-Methyl-pyridone-carboxamide Uric acid Dimethylguanosine Nitrosodimethylamine Uridine Erythritol Nitrosomethylamine Xanthine γ-Guanidinobutyrate Orotic acid Xanthosine | Indoxyl sulfate * p-Cresyl * Homocysteine * p-OH-hippurate 2-Methoxyresorcinol Indole-3-acetate Pentosidine 3-Deoxyglucosone Phenol CMPF Kinurenine Phenylacetic acid Fructoselysine Kinurenic acid Glyoxal Melatonin Putrescine Hippuric acid Methylglyoxal Quinolinic acid Nɛ-Carboxymethyllysine Spermidine Hydroquinone Spermine | β2-Microglobulin Adiponectin Dinucleoside polyphosphates Methionine-enkephalin Adrenomedullin DIP I Motiline Atrial natriuretic peptide δ-Sleep-inducing peptide Neuropeptide Y Endothelin Octopamine β-Endorphin Ghrelin Orexin A β-Lipotropin Hepcidin Parathyroid hormone Basic fibroblast growth factor Hyaluronic acid Retinol binding protein Calcitonin-gene related peptide | Interleukin-1β Interleukin-6 Tumor necrosis factor α Substance P Cholecystokinin Clara cell protein Interleukin-18 Up4A Complement factor D Resistin κ-Ig Light chain Uroguanylin Cystatin C λ-Ig Light chain Vasoactive intestinal peptide Desacylghrelin Leptin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lara-Prado, J.I.; Pazos-Pérez, F.; Méndez-Landa, C.E.; Grajales-García, D.P.; Feria-Ramírez, J.A.; Salazar-González, J.J.; Cruz-Romero, M.; Treviño-Becerra, A. Acute Kidney Injury and Organ Dysfunction: What Is the Role of Uremic Toxins? Toxins 2021, 13, 551. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080551
Lara-Prado JI, Pazos-Pérez F, Méndez-Landa CE, Grajales-García DP, Feria-Ramírez JA, Salazar-González JJ, Cruz-Romero M, Treviño-Becerra A. Acute Kidney Injury and Organ Dysfunction: What Is the Role of Uremic Toxins? Toxins. 2021; 13(8):551. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080551
Chicago/Turabian StyleLara-Prado, Jesús Iván, Fabiola Pazos-Pérez, Carlos Enrique Méndez-Landa, Dulce Paola Grajales-García, José Alfredo Feria-Ramírez, Juan José Salazar-González, Mario Cruz-Romero, and Alejandro Treviño-Becerra. 2021. "Acute Kidney Injury and Organ Dysfunction: What Is the Role of Uremic Toxins?" Toxins 13, no. 8: 551. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080551