The Allosteric Activation of α7 nAChR by α-Conotoxin MrIC Is Modified by Mutations at the Vestibular Site

 ,
, 
 , and
, and
Abstract
:1. Introduction
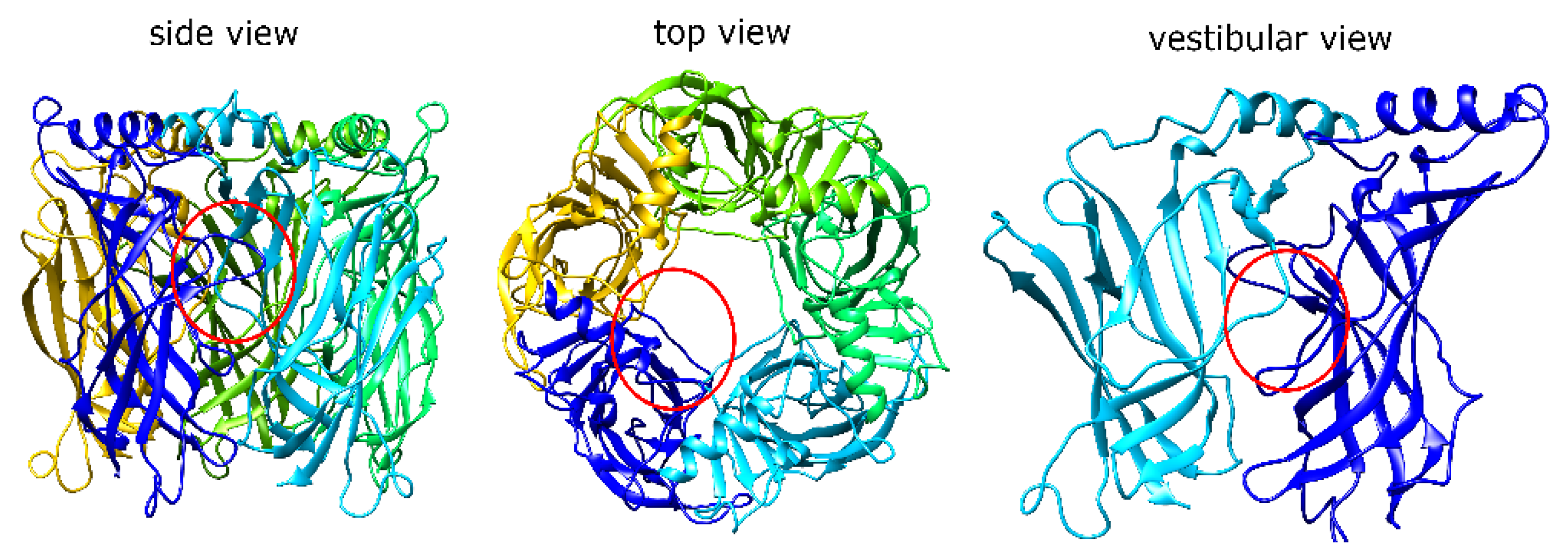
1.1. α7 nAChR Structure and Properties
1.2. α-Conotoxin Mr1.7 Variants Target α7 nAChR
1.3. α7 nAChR Activation by MrIC May Be through an Allosteric Binding Site
2. Results and Discussion
2.1. MrIA Failed to Bind in the AA Site but Bound Close to the Vestibular Pore
2.2. MrIB Shifted the Vestibular Loops Away from Each Other
2.3. MrIC Anchored the AA Site through Its N-Terminal Proline
2.4. MrIC Docked to the Orthosteric Site of α7 nAChR and Formed Several Hydrophobic Interactions
2.5. MrIC Binding to the Orthosteric Site Was Insignificant in Electrophysiology Experiments
2.6. Upon Co-Application with PNU-120596, MrIC Could Activate the α7C190 Mutant, Which Cannot Be Orthosterically Activated
2.7. MrIC Binding to the ECD AA Site Was Tested with Three AA Site Mutants
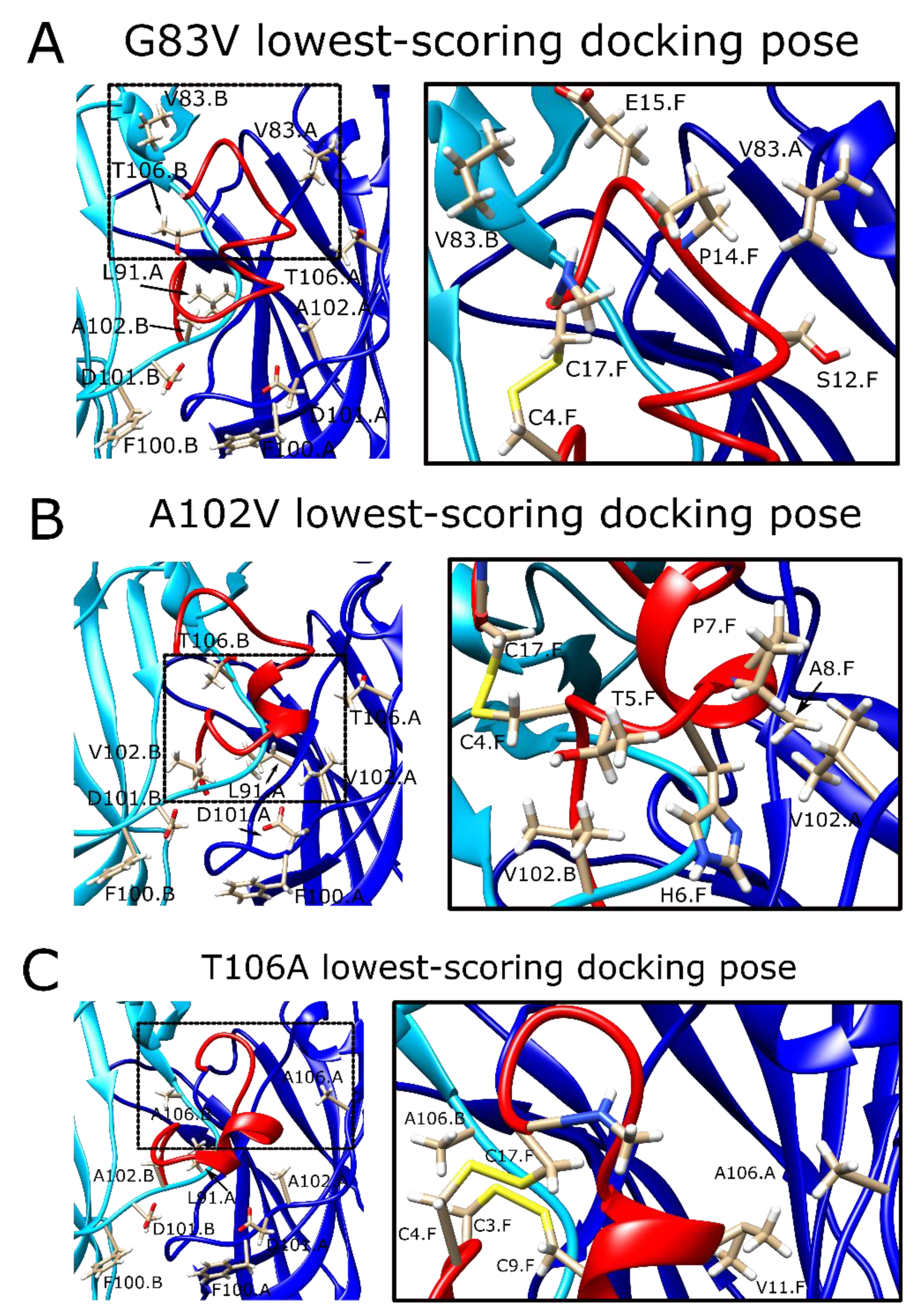
2.8. The α7G83V Mutation Caused an Increase in the Binding Strength of Several Peptide Residues in Peptide Docking Calculations
2.9. The α7A102V Mutation Improved the Binding Strength Slightly Less Than the α7G83V Mutation
2.10. α7T106A Peptide Docking Poses Were Similar to the WT α7 nAChR with Only Minor Improvement of Interaction Energy
2.11. α7G83V and α7A102V Mutations Caused Increased MrIC Responses Compared to WT α7 nAChR in Electrophysiology Experiments
2.12. The α7T106A Mutation Resulted in Background Responses to PNU-120596 with an Enhancement Caused by MrIC
2.13. Limitations of the Study
3. Conclusions
4. Materials and Methods
4.1. Homology Modeling of α7 ECD
4.2. Modeling of MrIA, MrIB, and MrIC
4.3. Docking of the Peptides into α7 nAChR Models
4.4. Chemicals and Reagents
4.5. Peptide Regioselective Synthesis and Purification
4.6. Heterologous Expression of nAChRs in Xenopus Laevis Oocytes
4.7. Two-Electrode Voltage-Clamp Electrophysiology
4.8. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Séguéla, P.; Wadiche, J.; Dineley-Miller, K.; Dani, J.A.; Patrick, J.W. Molecular cloning, functional properties, and distribution of rat brain α7: A nicotinic cation channel highly permeable to calcium. J. Neurosci. 1993, 13, 596–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, S.; Bertrand, D.; Matter, J.M.; Hernandez, M.C.; Bertrand, S.; Millar, N.; Valera, S.; Barkas, T.; Ballivet, M. A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTX. Neuron 1990, 5, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Bertrand, S.; Binzoni, T.; Bertrand, D. Neuronal nicotinic α7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 1996, 491, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Mose, T.N.; Blakeman, S.; Picciotto, M.R. Hippocampal α7 nicotinic ACh receptors contribute to modulation of depression-like behaviour in C57BL/6J mice. Br. J. Pharmacol. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Xu, X.; Pan, L.; Zhu, W.; Fu, X.; Guo, L.; Lu, Q.; Wang, J. Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress. J. Neuroinflamm. 2017, 14, 234. [Google Scholar] [CrossRef]
- Martin, L.F.; Freedman, R. Schizophrenia and the α7 Nicotinic Acetylcholine Receptor. In International Review of Neurobiology; Academic Press: Cambridge, MA, USA, 2007; Volume 78, pp. 225–246. ISBN 0123737370. [Google Scholar]
- Beinat, C.; Banister, S.D.; Herrera, M.; Law, V.; Kassiou, M. The therapeutic potential of α7 nicotinic acetylcholine receptor (α7 nAChR) agonists for the treatment of the cognitive deficits associated with schizophrenia. CNS Drugs 2015, 29, 529–542. [Google Scholar] [CrossRef]
- Wallace, T.L.; Ballard, T.M.; Pouzet, B.; Riedel, W.J.; Wettstein, J.G. Drug targets for cognitive enhancement in neuropsychiatric disorders. Pharmacol. Biochem. Behav. 2011, 99, 130–145. [Google Scholar] [CrossRef]
- Horenstein, N.A.; Papke, R.L. Anti-inflammatory silent agonists. ACS Med. Chem. Lett. 2017, 8, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Bagdas, D.; Gurun, M.S.; Flood, P.; Papke, R.L.; Damaj, M.I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on α7 nAChRs. Curr. Neuropharmacol. 2018, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, D.; Terry, A.V. The wonderland of neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 2018, 151, 214–225. [Google Scholar] [CrossRef]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox—Transforming natural peptides into peptide therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.D.; McIntosh, J.M.; Hillyard, D.R.; Cruz, L.J.; Olivera, B.M. The A-superfamily of conotoxins: Structural and functional divergence. J. Biol. Chem. 2004, 279, 17596–17606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giribaldi, J.; Dutertre, S. α-Conotoxins to explore the molecular, physiological and pathophysiological functions of neuronal nicotinic acetylcholine receptors. Neurosci. Lett. 2018, 679, 24–34. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Liu, N.; Wu, C.; Jiang, J.; Yue, J.; Jing, Y.; Dai, Q. Diversity and evolution of conotoxins in Conus virgo, Conus eburneus, Conus imperialis and Conus marmoreus from the South China Sea. Toxicon 2012, 60, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Vetter, I.; Dutertre, S.; Abraham, N.; Emidio, N.B.; Inserra, M.; Murali, S.S.; Christie, M.J.; Alewood, P.F.; Lewis, R.J. MrIC, a novel α-conotoxin agonist in the presence of PNU at endogenous α7 nicotinic acetylcholine receptors. Biochemistry 2014, 53, 1–3. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Brust, A.; Jin, A.H.; Alewood, P.F.; Dutertre, S.; Lewis, R.J. Cone snail venomics: From novel biology to novel therapeutics. Future Med. Chem. 2014, 6, 1659–1675. [Google Scholar] [CrossRef]
- Mueller, A.; Starobova, H.; Inserra, M.C.; Jin, A.H.; Deuis, J.R.; Dutertre, S.; Lewis, R.J.; Alewood, P.F.; Daly, N.L.; Vetter, I. α-conotoxin MrIC is a biased agonist at α7 nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 94, 155–163. [Google Scholar] [CrossRef]
- Williams, D.K.; Wang, J.; Papke, R.L. Investigation of the Molecular Mechanism of the α7 Nicotinic Acetylcholine Receptor Positive Allosteric Modulator PNU-120596 Provides Evidence for Two Distinct Desensitized States. Mol. Pharmacol. 2011, 80, 1013–1032. [Google Scholar] [CrossRef]
- Gulsevin, A.; Papke, R.L.; Stokes, C.; Garai, S.; Thakur, G.A.; Quadri, M.; Horenstein, N.A. Allosteric agonism of α7 nicotinic acetylcholine receptors: Receptor modulation outside the orthosteric site. Mol. Pharmacol. 2019, 95, 606–614. [Google Scholar] [CrossRef]
- Hurst, R.S.; Hajós, M.; Raggenbass, M.; Wall, T.M.; Higdon, N.R.; Lawson, J.A.; Rutherford-Root, K.L.; Berkenpas, M.B.; Hoffmann, W.E.; Piotrowski, D.W.; et al. A Novel Positive Allosteric Modulator of the α7 Neuronal Nicotinic Acetylcholine Receptor: In Vitro and In Vivo Characterization. J. Neurosci. 2005, 25, 4396–4405. [Google Scholar] [CrossRef] [Green Version]
- Gulsevin, A. Nicotinic receptor pharmacology in silico: Insights and challenges. Neuropharmacology 2020, 177, 108257. [Google Scholar] [CrossRef]
- Horenstein, N.A.; Papke, R.L.; Kulkarni, A.R.; Chaturbhuj, G.U.; Stokes, C.; Manther, K.; Thakur, G.A. Critical molecular determinants of α7 nicotinic acetylcholine receptor allosteric activation: Separation of direct allosteric activation and positive allosteric modulation. J. Biol. Chem. 2016, 291, 5049–5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadri, M.; Garai, S.; Thakur, G.A.; Stokes, C.; Gulsevin, A.; Horenstein, N.A.; Papke, R.L. Macroscopic and microscopic activation of α7 nicotinic acetylcholine receptors by the structurally unrelated allosteric agonist-positive allosteric modulators (ago-PAMs) B-973B and GAT107. Mol. Pharmacol. 2019, 95, 43–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papke, R.L.; Garai, S.; Stokes, C.; Horenstein, N.A.; Zimmerman, A.D.; Abboud, K.A.; Thakur, G.A. Differing activity profiles of the stereoisomers of 2,3,5,6TMP-TQS, a putative silent allosteric modulator of α7nAChR. Mol. Pharmacol. 2020, 98, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [Green Version]
- Gulsevin, A.; Meiler, J. An investigation of three-finger toxin-nachr interactions through rosetta protein docking. Toxins 2020, 12, 598. [Google Scholar] [CrossRef]
- Papke, R.L.; Porter Papke, J.K. Comparative pharmacology of rat and human α7 nAChR conducted with net charge analysis. Br. J. Pharmacol. 2002, 137, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.K.; Dhankher, P.; Sheppard, T.D.; Sher, E.; Millar, N.S. A series of α7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol. Pharmacol. 2012, 81, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Thakur, G.A.; Kulkarni, A.R.; Deschamps, J.R.; Papke, R.L. Expeditious synthesis, enantiomeric resolution, and enantiomer functional characterization of (4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (4BP-TQS): An allosteric agonist-positive allosteric modulator of α7 nicotinic ac. J. Med. Chem. 2013, 56, 8943–8947. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.K.; Savolainen, M.; Young, G.T.; Zwart, R.; Sher, E.; Millar, N.S. Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. USA 2011, 108, 5867–5872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAchR by α-Conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurny, R.; Debaveye, S.; Farinha, A.; Veys, K.; Vos, A.M.; Gossas, T.; Atack, J.; Bertrand, S.; Bertrand, D.; Danielson, U.H.; et al. Molecular blueprint of allosteric binding sites in a homologue of the agonist-binding domain of the α7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2015, 112, E2543–E2552. [Google Scholar] [CrossRef] [Green Version]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bradley, P.; Greisen, P.; Liu, Y.; Mulligan, V.K.; Kim, D.E.; Baker, D.; DiMaio, F. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J. Chem. Theory Comput. 2016, 12, 6201–6212. [Google Scholar] [CrossRef]
- Li, S.-X.; Huang, S.; Bren, N.; Noridomi, K.; Dellisanti, C.D.; Sine, S.M.; Chen, L. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat. Neurosci. 2011, 14, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.-W.; Kim, D.-H.; Olivera, B.M.; McIntosh, J.M.; Han, K.-H. Solution conformation of a neuronal nicotinic acetylcholine receptor antagonist α-conotoxin OmIA that discriminates α3 vs. α6 nAChR subtypes. Biochem. Biophys. Res. Commun. 2006, 345, 248–254. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Leaver-Fay, A.; O’Meara, M.J.; Tyka, M.; Jacak, R.; Song, Y.; Kellogg, E.H.; Thompson, J.; Davis, I.W.; Pache, R.A.; Lyskov, S.; et al. Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol. 2013, 523, 109–143. [Google Scholar] [PubMed] [Green Version]
- Tyka, M.D.; Keedy, D.A.; André, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins Struct. Funct. Bioinform. 2010, 78, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.R.; Thakur, G.A. Microwave-assisted expeditious and efficient synthesis of cyclopentene ring-fused tetrahydroquinoline derivatives using three-component Povarov reaction. Tetrahedron Lett. 2013, 54, 6592–6595. [Google Scholar] [CrossRef] [Green Version]
- Jin, A.H.; Dekan, Z.; Smout, M.J.; Wilson, D.; Dutertre, S.; Vetter, I.; Lewis, R.J.; Loukas, A.; Daly, N.L.; Alewood, P.F. Conotoxin Φ-MiXXVIIA from the Superfamily G2 Employs a Novel Cysteine Framework that Mimics Granulin and Displays Anti-Apoptotic Activity. Angew. Chem. Int. Ed. 2017, 56, 14973–14976. [Google Scholar] [CrossRef]
- Halevi, S.; Yassin, L.; Eshel, M.; Sala, F.; Sala, S.; Criado, M.; Treinin, M. Conservation within the RIC-3 Gene Family: Effectors of Mammalian Nicotinic Acetylcholine Receptor Expression. J. Biol. Chem. 2003, 278, 34411–34417. [Google Scholar] [CrossRef] [Green Version]
- Papke, R.L.; Stokes, C.; Williams, D.K.; Wang, J.; Horenstein, N.A. Cysteine accessibility analysis of the human alpha7 nicotinic acetylcholine receptor ligand-binding domain identifies L119 as a gatekeeper. Neuropharmacology 2011, 60, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Papke, R.L.; Stokes, C. Working with OpusXpress: Methods for high volume oocyte experiments. Methods 2010, 51, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Williams, D.K.; Stokes, C.; Horenstein, N.A.; Papke, R.L. The effective opening of nicotinic acetylcholine receptors with single agonist binding sites. J. Gen. Physiol. 2011, 137, 369–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, C.; Garai, S.; Kulkarni, A.R.; Cantwell, L.N.; Noviello, C.M.; Hibbs, R.E.; Horenstein, N.A.; Abboud, K.A.; Thakur, G.A.; Papke, R.L. Heteromeric neuronal nicotinic acetylcholine receptors with mutant B subunits acquire sensitivity to A7-selective positive allosteric modulatorss. J. Pharmacol. Exp. Ther. 2019, 370, 252–268. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence |
|---|---|
| MrIA | ECCTHPACHVSNPELC-NH2 |
| MrIB | ROECCTHOACHVSNPELCS-OH |
| MrIC | PECCTHPACHVSNPELC-NH2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulsevin, A.; Papke, R.L.; Stokes, C.; Tran, H.N.T.; Jin, A.H.; Vetter, I.; Meiler, J. The Allosteric Activation of α7 nAChR by α-Conotoxin MrIC Is Modified by Mutations at the Vestibular Site. Toxins 2021, 13, 555. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080555
Gulsevin A, Papke RL, Stokes C, Tran HNT, Jin AH, Vetter I, Meiler J. The Allosteric Activation of α7 nAChR by α-Conotoxin MrIC Is Modified by Mutations at the Vestibular Site. Toxins. 2021; 13(8):555. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080555
Chicago/Turabian StyleGulsevin, Alican, Roger L. Papke, Clare Stokes, Hue N. T. Tran, Aihua H. Jin, Irina Vetter, and Jens Meiler. 2021. "The Allosteric Activation of α7 nAChR by α-Conotoxin MrIC Is Modified by Mutations at the Vestibular Site" Toxins 13, no. 8: 555. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080555