PKCζ-Mitogen-Activated Protein Kinase Signaling Mediates Crotalphine-Induced Antinociception

 , ,
, ,
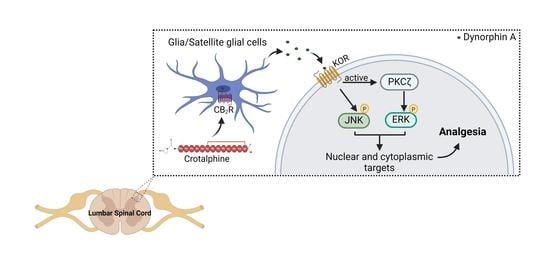
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
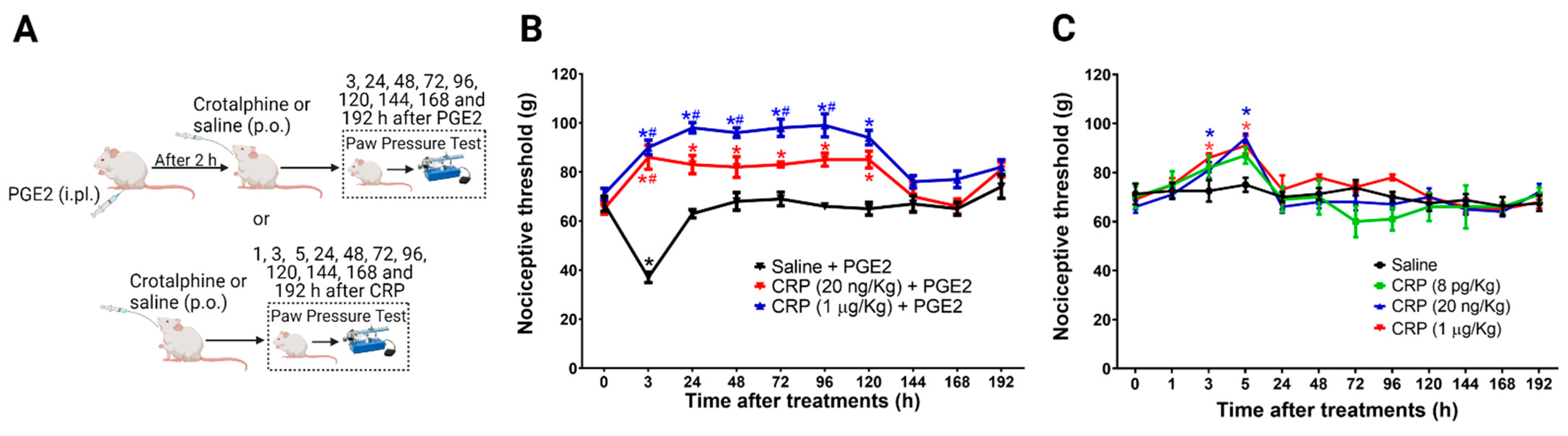
2.1. PGE2 Is Responsible for the Long-Lasting Antinociceptive Effect Induced by Crotalphine
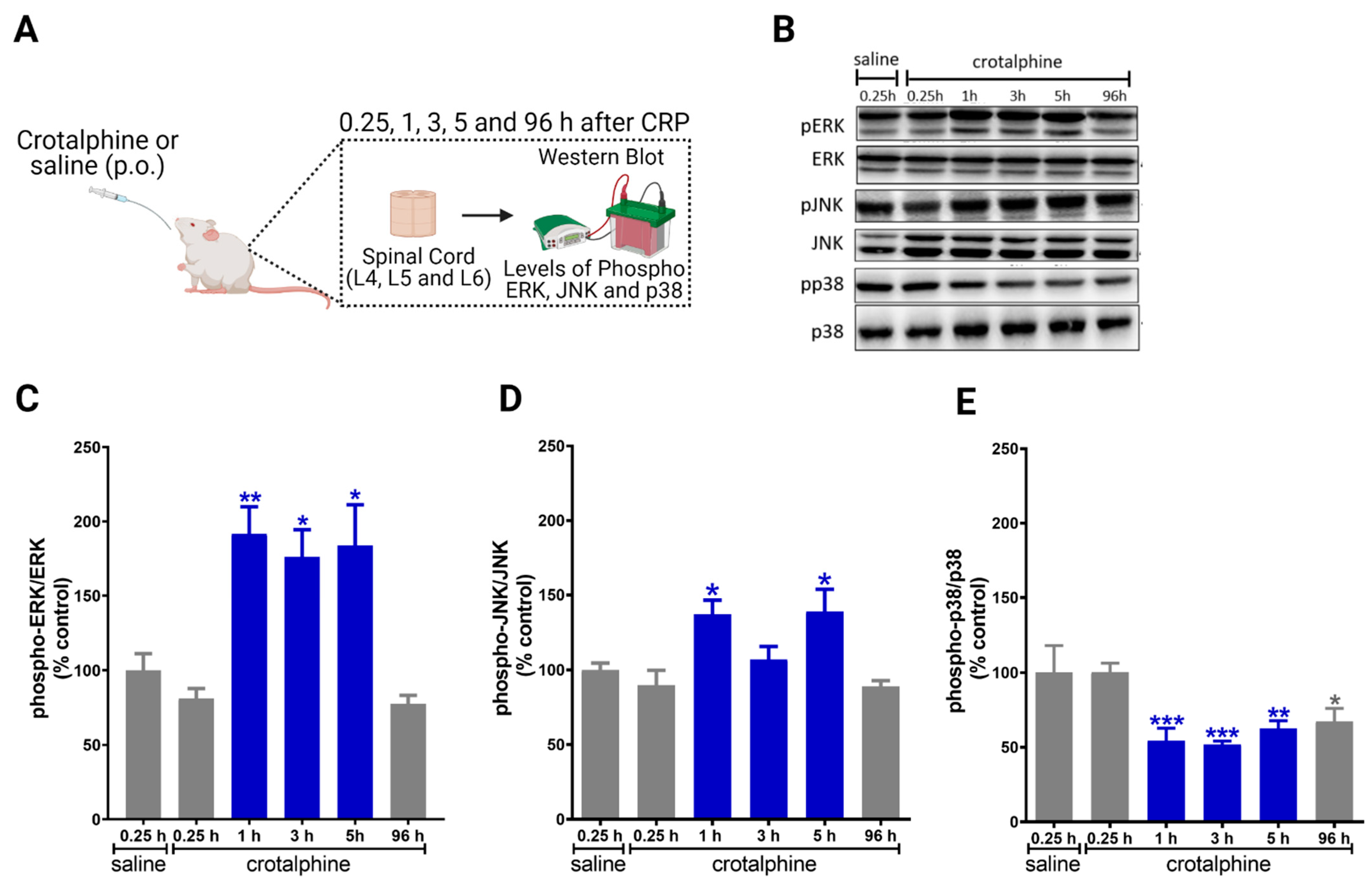
2.2. Crotalphine Increases the Spinal ERK and JNK and Decreases p38 Activation
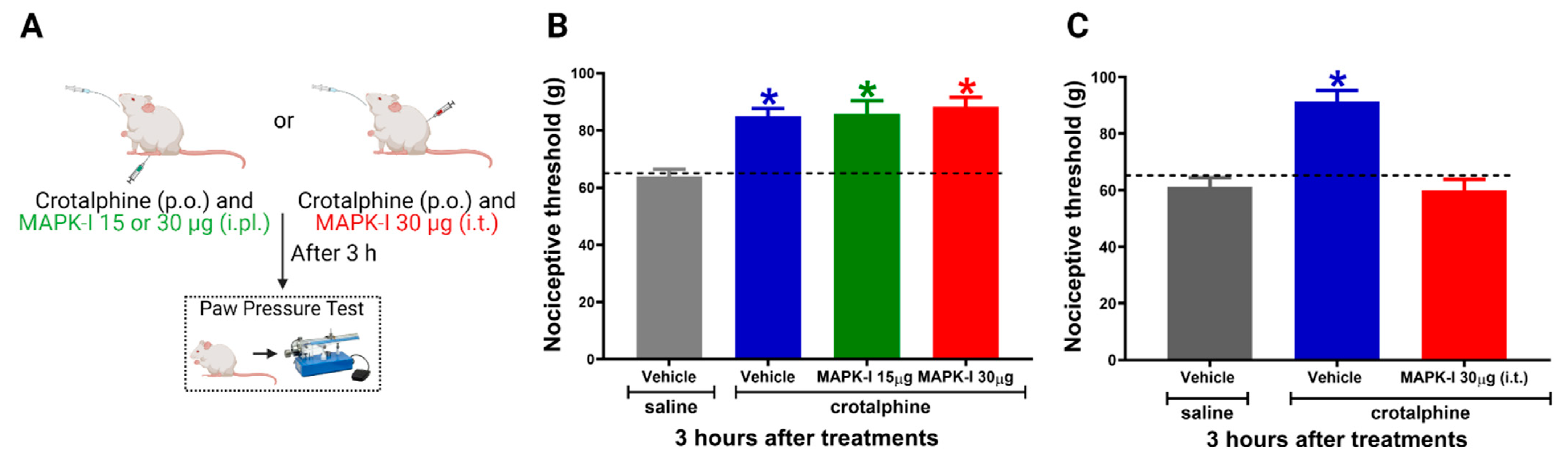
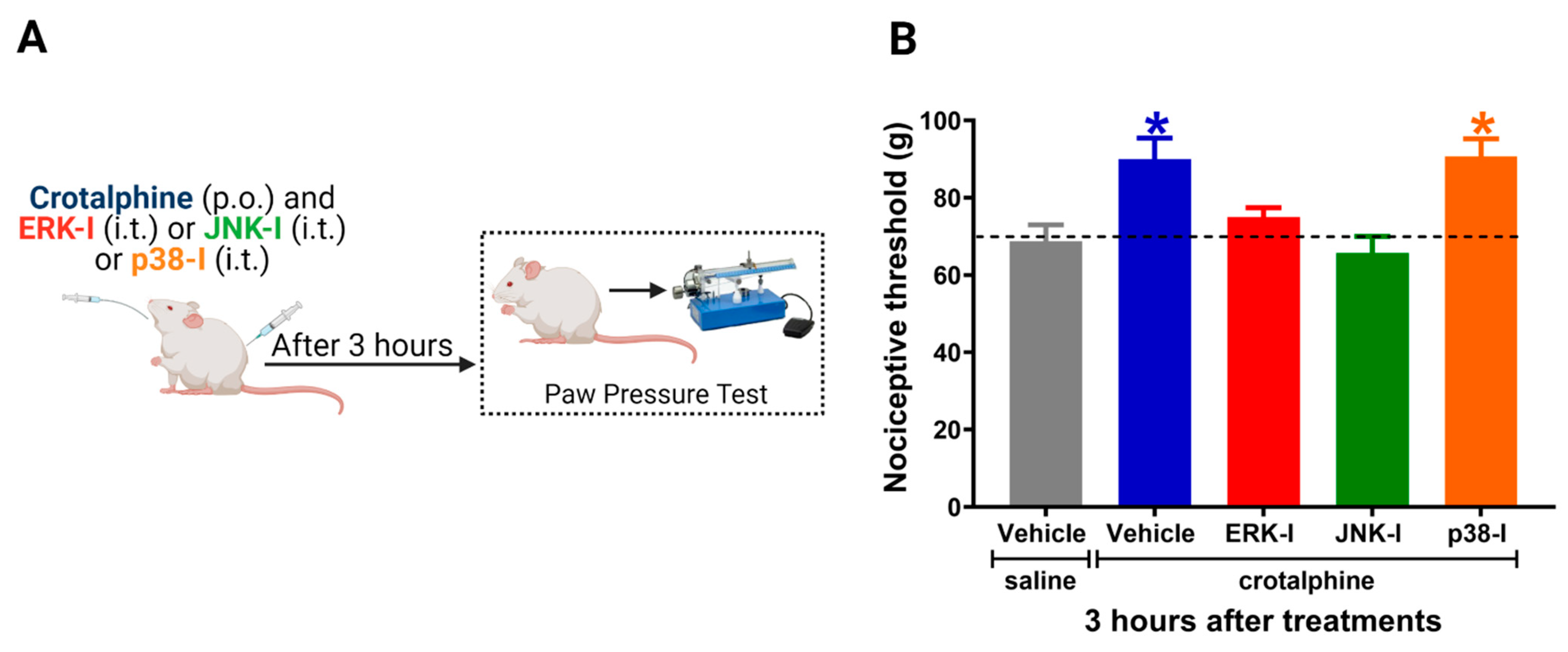
2.3. Spinal ERK and JNK Activation Participates in Crotalphine-Induced Analgesia
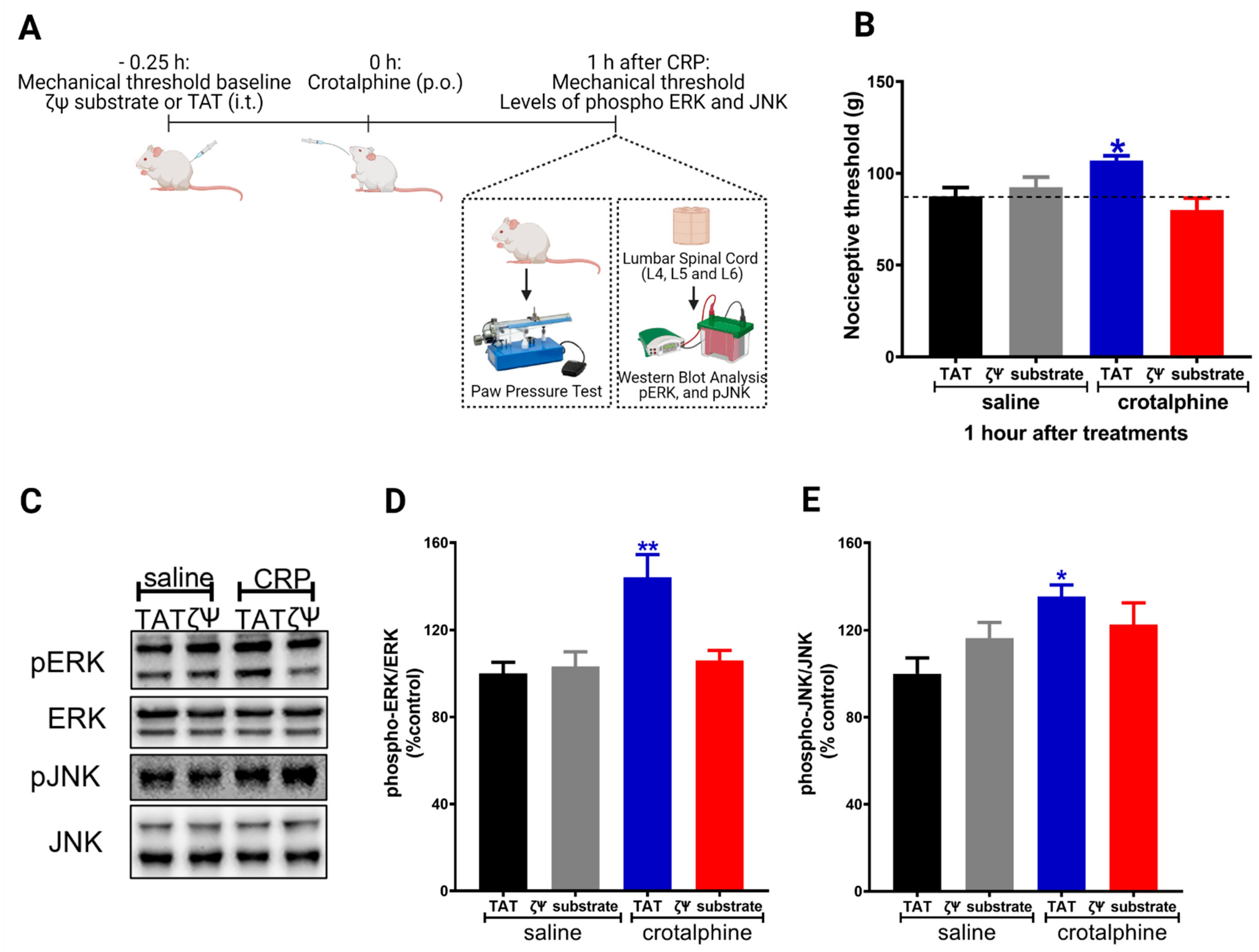
2.4. Spinal PKCζ Is Involved in the Antinociceptive Effect of Crotalphine
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Chemicals and DrugAadministration
5.3. Behavioral Assessment
Evaluation of the Antinociceptive Effect
5.4. Biochemical Studies
Western Blot Analysis
5.5. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Konno, K.; Picolo, G.; Gutierrez, V.P.; Brigatte, P.; Zambelli, V.O.; Camargo, A.C.M.; Cury, Y. Crotalphine, a novel potent analgesic peptide from the venom of the South American rattlesnake Crotalus durissus terrificus. Peptides 2008, 29, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Machado, F.C.; Zambelli, V.O.; Fernandes, A.C.O.; Heimann, A.S.; Cury, Y.; Picolo, G. Peripheral interactions between cannabinoid and opioid systems contribute to the antinociceptive effect of crotalphine. Br. J. Pharmacol. 2014, 171, 961–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressan, E.; Touska, F.; Vetter, I.; Kistner, K.; Kichko, T.I.; Teixeira, N.B.; Picolo, G.; Cury, Y.; Lewis, R.J.; Fischer, M.J.M.; et al. Crotalphine desensitizes TRPA1 ion channels to alleviate inflammatory hyperalgesia. Pain 2016, 157, 2504–2516. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Obata, K.; Noguchi, K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004, 74, 2643–2653. [Google Scholar] [CrossRef]
- Abraham, A.D.; Schattauer, S.S.; Reichard, K.L.; Cohen, J.H.; Fontaine, H.M.; Song, A.J.; Johnson, S.D.; Land, B.B.; Chavkin, C. Estrogen regulation of GRK2 inactivates Kappa opioid receptor signaling mediating analgesia, but not aversion. J. Neurosci. 2018, 38, 8031–8043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Freitas, B.G.; Pereira, L.M.; Santa-Cecília, F.V.; Hösch, N.G.; Picolo, G.; Cury, Y.; Zambelli, V.O. Mitogen-Activated Protein Kinase Signaling Mediates Morphine Induced-Delayed Hyperalgesia. Front. Neurosci. 2019, 13, 1018. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, V.O.; Fernandes, A.C.D.O.; Gutierrez, V.P.; Ferreira, J.C.B.; Parada, C.A.; Mochly-Rosen, D.; Cury, Y. Peripheral sensitization increases opioid receptor expression and activation by crotalphine in rats. PLoS ONE 2014, 9, e90576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, M.M.; Clark, A.L.; Haas, P.D.; Serna, J.S.; Hahn, J.W.; Kiss, A.; Coscia, C.J. μ and κ opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J. Biol. Chem. 2005, 280, 27662–27669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berra, E.; Diaz-Meco, M.T.; Dominguez, I.; Municio, M.M.; Sanz, L.; Lozano, J.; Chapkin, R.S.; Moscat, J. Protein kinase C ζ isoform is critical for mitogenic signal transduction. Cell 1993, 74, 555–563. [Google Scholar] [CrossRef]
- He, Y.; Wang, Z.J. Nociceptor beta II, delta, and epsilon isoforms of PKC differentially mediate paclitaxel-induced spontaneous and evoked pain. J. Neurosci. 2015, 35, 4614–4625. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, V.P.; Konno, K.; Chacur, M.; Sampaio, S.C.; Picolo, G.; Brigatte, P.; Zambelli, V.O.; Cury, Y. Crotalphine induces potent antinociception in neuropathic pain by acting at peripheral opioid receptors. Eur. J. Pharmacol. 2008, 594, 84–92. [Google Scholar] [CrossRef]
- Gutierrez, V.P.; Zambelli, V.O.; Picolo, G.; Chacur, M.; Sampaio, S.C.; Brigatte, P.; Konno, K.; Cury, Y. The peripheral L-arginine-nitric oxide-cyclic GMP pathway and ATP-sensitive K+ channels are involved in the antinociceptive effect of crotalphine on neuropathic pain in rats. Behav. Pharmacol. 2012, 23, 14–24. [Google Scholar] [CrossRef]
- Van Elstraete, A.C.; Sitbon, P.; Trabold, F.; Mazoit, J.X.; Benhamou, D. A single dose of intrathecal morphine in rats induces long-lasting hyperalgesia: The protective effect of prior administration of ketamine. Anesth. Analg. 2005, 101, 1750–1756. [Google Scholar] [CrossRef]
- Bohn, L.M.; Belcheva, M.M.; Coscia, C.J. Mitogenic signaling via endogenous κ-opioid receptors in C6 glioma cells: Evidence for the involvement of protein kinase C and the mitogen- activated protein kinase signaling cascade. J. Neurochem. 2000, 74, 564–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchas, M.R.; Macey, T.A.; Lowe, J.D.; Chavkin, C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.B.; Lonsdorf, T.B.; Schalling, M.; Kosek, E.; Ingvar, M. Increased sensitivity to thermal pain following a single opiate dose is influenced by the COMT val158 met polymorphism. PLoS ONE 2009, 4, e6016. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.Y.; Wen, Y.R.; Zhang, D.R.; Borsello, T.; Bonny, C.; Strichartz, G.R.; Decosterd, I.; Ji, R.R. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: Respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J. Neurosci. 2006, 26, 3551–3560. [Google Scholar] [CrossRef] [Green Version]
- Minden, A.; Lin, A.; Smeal, T.; Dérijard, B.; Cobb, M.; Davis, R.; Karin, M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol. Cell. Biol. 1994, 14, 6683–6688. [Google Scholar] [CrossRef]
- Ostenfeld, T.; Krishen, A.; Lai, R.Y.; Bullman, J.; Green, J.; Anand, P.; Scholz, J.; Kelly, M. A randomized, placebo-controlled trial of the analgesic efficacy and safety of the p38 MAP kinase inhibitor, losmapimod, in patients with neuropathic pain from lumbosacral radiculopathy. Clin. J. Pain 2015, 31, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Espírito Santo, V.; Passos, J.; Nzwalo, H.; Carvalho, I.; Santos, F.; Martins, C.; Salgado, L.; Silva, C.E.; Vinhais, S.; Vilares, M.; et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1: A single-institution experience. J. Neurooncol. 2020, 147, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.E.; Daniels, S.E.; Black, P.; Chang, S.; Protter, A.; Desjardins, P.J. Novel p38α mitogen-activated protein kinase inhibitor shows analgesic efficacy in acute postsurgical dental pain. J. Clin. Pharmacol. 2012, 52, 717–728. [Google Scholar] [CrossRef]
- Bruchas, M.R.; Yang, T.; Schreiber, S.; DeFino, M.; Kwan, S.C.; Li, S.; Chavkin, C. Long-acting κ opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J. Biol. Chem. 2007, 282, 29803–29811. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Sakurada, S.; Kohno, K.; Shiohira, H.; Katsuyama, S.; Sakurada, C.; Tsuzuki, M.; Sakurada, T. Spinal ERK activation via NO-cGMP pathway contributes to nociceptive behavior induced by morphine-3-glucuronide. Biochem. Pharmacol. 2009, 78, 1026–1034. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Schleicher, S.; Butcher, R.D.; Craig, A.; Lieb, K. The Neuropeptide Substance P Activates p38 Mitogen-Activated Protein Kinase Resulting in IL-6 Expression Independently from NF-κB. J. Immunol. 2000, 165, 5606–5611. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Gereau IV, R.W.; Malcangio, M.; Strichartz, G.R. MAP kinase and pain. Brain Res. Rev. 2009, 60, 135–148. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, N.; Liu, F.; Lin, J.; Liu, M.; Huang, C.; Liao, D.; Zhou, C.; Wang, H.; Shen, J. Activation of mitogen-activated protein kinases in satellite glial cells of the trigeminal ganglion contributes to substance P-mediated inflammatory pain. Int. J. Oral Sci. 2019, 11, 24. [Google Scholar] [CrossRef]
- Belcheva, M.M.; Szùcs, M.; Wang, D.; Sadee, W.; Coscia, C.J. μ-Opioid Receptor-mediated ERK Activation Involves Calmodulin-dependent Epidermal Growth Factor Receptor Transactivation. J. Biol. Chem. 2001, 276, 33847–33853. [Google Scholar] [CrossRef] [Green Version]
- Kam, A.Y.F.; Chan, A.S.L.; Wong, Y.H. Phosphatidylinositol-3 kinase is distinctively required for μ-, but not κ-opioid receptor-induced activation of c-Jun N-terminal kinase. J. Neurochem. 2004, 89, 391–402. [Google Scholar] [CrossRef]
- Kreibich, A.S.; Blendy, J.A. cAMP response element-binding protein is required for stress but not cocaine-induced reinstatement. J. Neurosci. 2004, 24, 6686–6692. [Google Scholar] [CrossRef] [Green Version]
- Carlezon, W.A.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Ma, W.; Zheng, W.H.; Powell, K.; Jhamandas, K.; Quirion, R. Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: An in vitro and in vivo study. Eur. J. Neurosci. 2001, 14, 1091–1104. [Google Scholar] [CrossRef]
- Milligan, E.D.; Twining, C.; Chacur, M.; Biedenkapp, J.; O’Connor, K.; Poole, S.; Tracey, K.; Martin, D.; Maier, S.F.; Watkins, L.R. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J. Neurosci. 2003, 23, 1026–1040. [Google Scholar] [CrossRef]
- Randall, L.O.; Selitto, J.J. A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 1957, 111, 409–419. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Freitas, B.G.; Hösch, N.G.; Pereira, L.M.; Barbosa, T.C.; Picolo, G.; Cury, Y.; Zambelli, V.O. PKCζ-Mitogen-Activated Protein Kinase Signaling Mediates Crotalphine-Induced Antinociception. Toxins 2021, 13, 912. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13120912
de Freitas BG, Hösch NG, Pereira LM, Barbosa TC, Picolo G, Cury Y, Zambelli VO. PKCζ-Mitogen-Activated Protein Kinase Signaling Mediates Crotalphine-Induced Antinociception. Toxins. 2021; 13(12):912. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13120912
Chicago/Turabian Stylede Freitas, Bárbara G., Natália G. Hösch, Leandro M. Pereira, Tereza C. Barbosa, Gisele Picolo, Yara Cury, and Vanessa O. Zambelli. 2021. "PKCζ-Mitogen-Activated Protein Kinase Signaling Mediates Crotalphine-Induced Antinociception" Toxins 13, no. 12: 912. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13120912