Development of an Electrochemical Immunosensor for Fumonisins Detection in Foods


Abstract
:1. Introduction
2. Results and Discussion
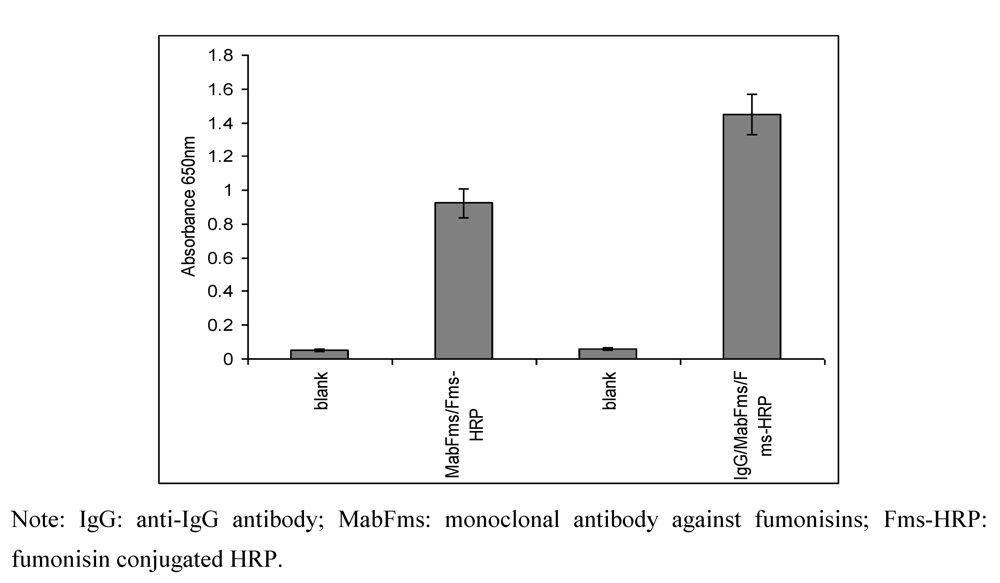
2.1. Optimization of the ELISA method

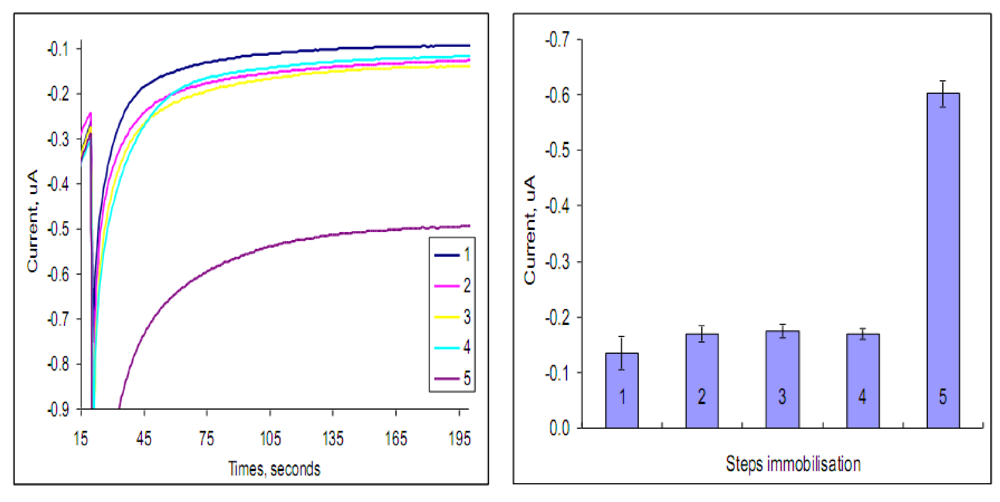
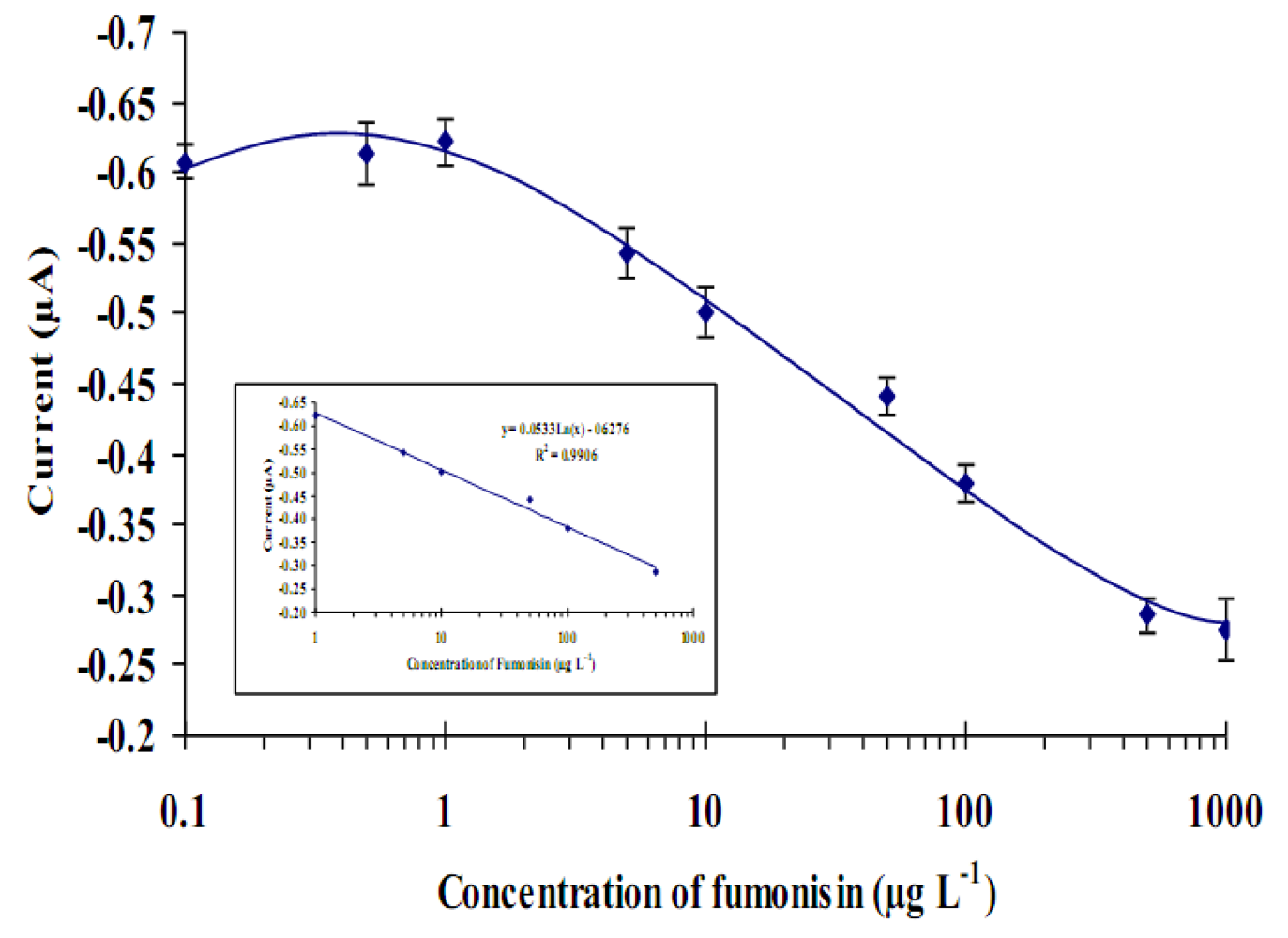
2.2. Optimization of the immunosensor




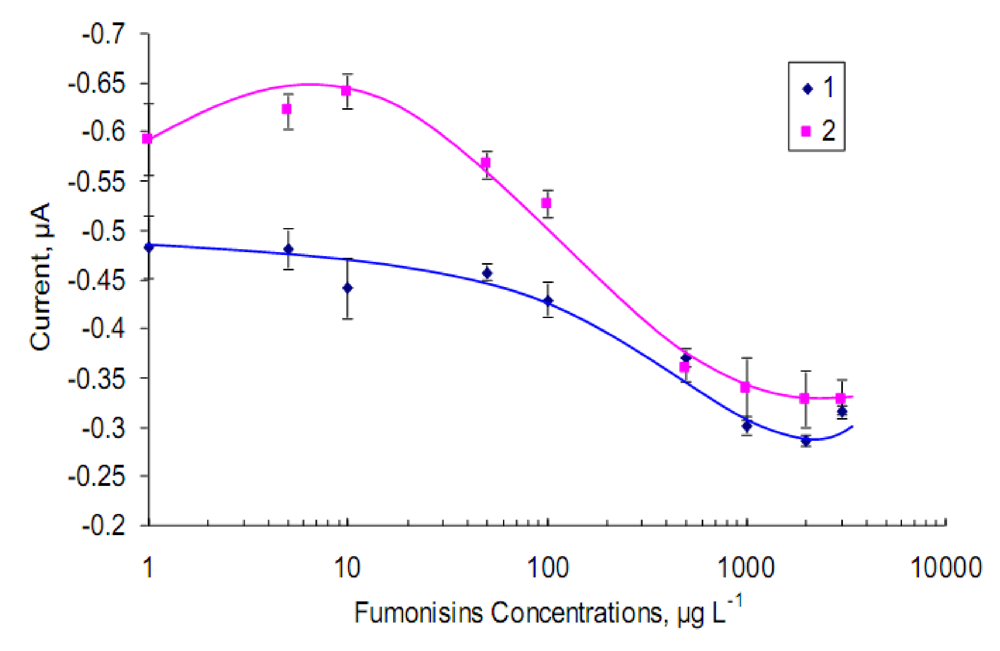
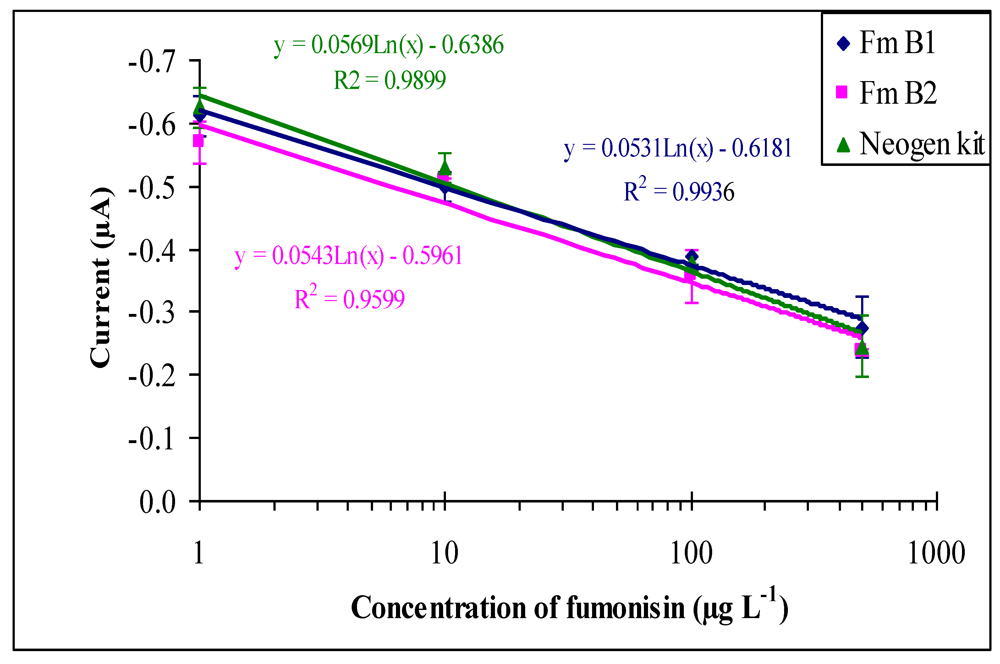
2.3. Cross-reactivity study

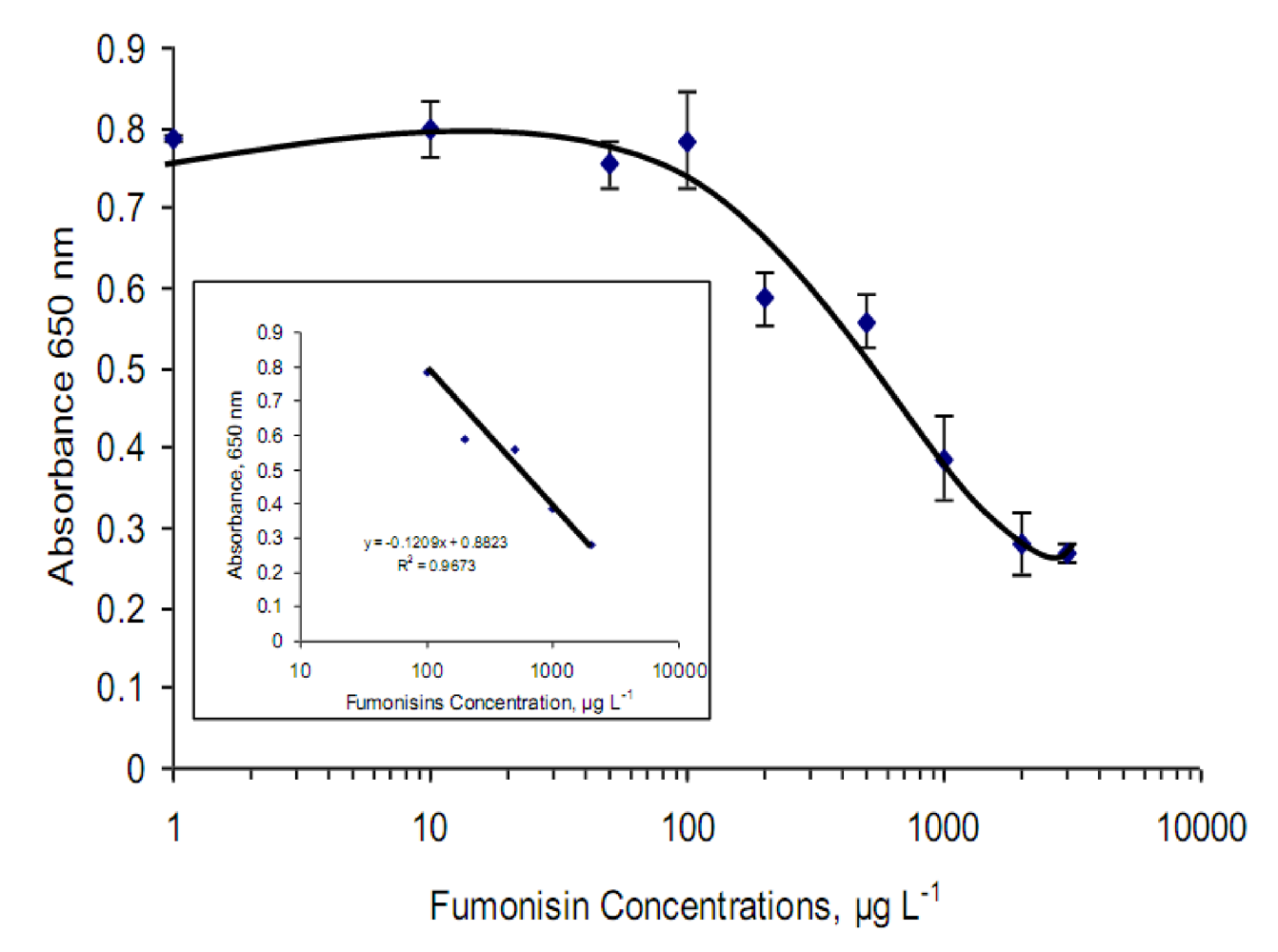
2.4. Optimized immunosensor calibration curve

2.5. Sensor response in extracted corn samples

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spiked Corn (µg·kg−1) | Without clean-up | Clean-up using C-18 | ||||
|---|---|---|---|---|---|---|
| Found (µg·kg−1) | % R | SD | Found (µg·kg−1) | % R | SD | |
| Non Spiked | <1 | 6.12 | <1 | 4.08 | ||
| 50 | 25.05 | 60.1 | 9.43 | 50.85 | 101.7 | 7.6 |
| 250 | 144.65 | 69.86 | 5.54 | 190.1 | 76.04 | 8.49 |
| 500 | 410.9 | 82.18 | 5.96 | 560.3 | 112.06 | 4.61 |
| 2500 | 2055.8 | 82.23 | 3.61 | 2616.05 | 104.64 | 3.87 |
3. Experimental Section
3.1. Reagents and solutions
3.2. Fabrication of screen-printed gold electrodes
3.3. Immunoassay developments
3.3.1. Direct immunoassay test
3.3.2. Competitive assay
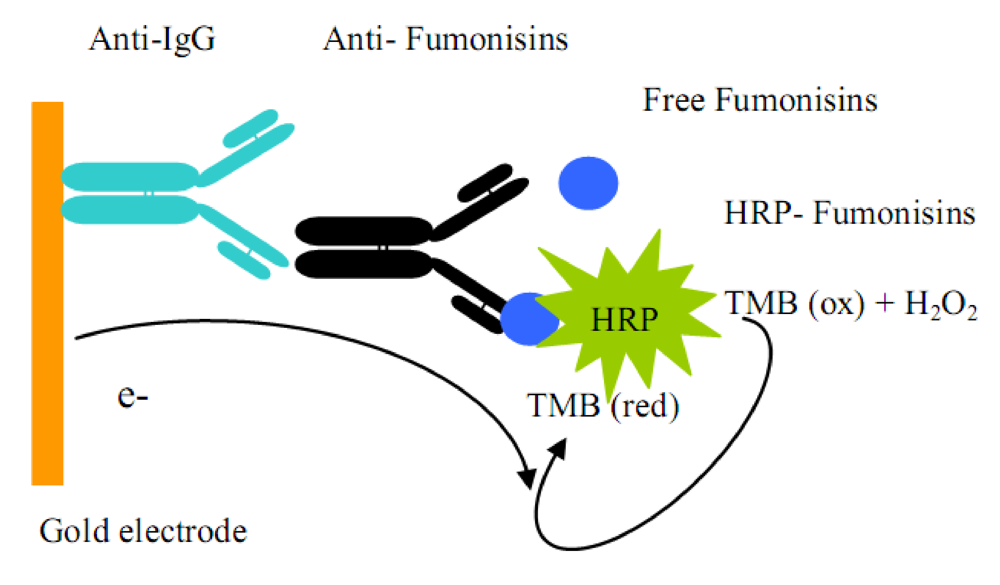
3.4. Electrochemical immunosensor
3.4.1. Electrochemical measurements
3.4.2. Direct competitive assay
3.4.3. Calibration plot and interpretation of result
3.5. Corn samples analysis
3.5.1. Extraction without clean-up
3.5.2. Clean up using C-18 solid phase extraction (SPE)
4. Conclusions
Acknowledgements
References and Notes
- Gelderblom, W.C.A.; Jaskiewicz, K.; Marasas, W.F.O.; Thiel, P.G.; Horak, R.M.; Vleggaar, R.; Kriek, N.P.J. Fumonisins-Novel mycotoxins with cancer-promoting activity produced by Fusarium moniliforme. Appl. Environ. Microbiol. 1988, 54, 1806–1811. [Google Scholar] [PubMed]
- Dutton, M.F. Fumonisins, mycotoxin of increasing importance: Their nature and their effects. Phramatcol. Ther. 1996, 70, 137–161. [Google Scholar] [CrossRef]
- Shepard, G.S.; Thiel, P.G.; Stockenstorom, S.; Sydenham, E.W. Worldwide survey of fumonisin contamination of corn and corn-based products. J. AOAC Int. 1996, 79, 671–787. [Google Scholar] [PubMed]
- Rheeder, J.P.; Marasas, W.F.O.; Vismer, H.F. Production of fumonisin analogs by Fusarium species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar] [PubMed]
- Jackson, L.; Jablonski, J. Fumonisins. In Mycotoxins in Food, Detection and Control; Magan, N., Olsen, M., Eds.; Woodhead Publishing Ltd.: Cambridge, UK, 2004; pp. 367–405. [Google Scholar]
- Marasas, W.F.O. Discovery and occurrence of the fumonisin: A historical perspective. Environ. Health Perspect. 2001, 109, 239–243. [Google Scholar]
- Colvin, B.M.; Harrison, L.R. Fumonisins induced pulmonary edema and hydrothorax in swine. Mycopathologia 1992, 117, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.M.; Silva, L.J.G.; Pena, A.L.S.; Silveira, M.I. Determination of fumonisins B1 and B2 in Portuguese maize and maize-based samples by HPLC with fluorescence detection. Anal. Bioanal. Chem. 2006, 384, 1214–1220. [Google Scholar] [PubMed]
- Rheeder, J.P.; Marasas, W.F.O.; Thiel, P.G.; Sydenham, E.W.; Sherpard, G.S.; van Schalkwyk, D.J. Fusarium moniliforme and fumonisins in corn in relation to human esophagaea cancer in Transkei. Phytopathology 1992, 82, 353–357. [Google Scholar]
- Pagliuca, G.; Zironi, E.; Ceccolini, A.; Matera, R.; Serrazanetti, P.G.; Piva, A. Simple method for the simultaneous isolation and determination of fumonisin B1 and its metabolite aminopentol-1 in swine liver by liquid chromatography–fluorescence detection. J. Chrom. B 2005, 819, 97–103. [Google Scholar]
- Wang, S.; Quan, Y.; Lee, N.; Kennedy, R.I. Rapid determination of fumonisin B1 in food samples by enzyme-linked immunosorbent assay and colloid gold immunoassay. J. Agric. Food Chem. 2006, 54, 2491–2495. [Google Scholar] [PubMed]
- Shelby, R.A.; Rottinghaus, G.E.; Minor, H.C. Comparison of thin layer chromatography and competitive immunoassay methods for detecting fumonisins on maize. J. Agric. Food Chem. 1994, 42, 2064–2067. [Google Scholar] [CrossRef]
- Sydenham, E.W.; Shephard, G.S. Chromatographic and allied methods of analysisfor selected mycotoxins. In Progress in Food Contaminant Analysis; Gilbert, J., Ed.; Blackie Academic & Professional: London, UK, 1996; pp. 65–146. [Google Scholar]
- Shepard, G.S. Chromatographic determination of the fumonisins mycotoxins. J. Chrom. A 1998, 815, 31–39. [Google Scholar]
- Schneider, E.; Usleber, E.; Märtlbauer, E. Rapid detection of fumonisin B1 in corn-based food by competitive direct dipstick enzyme immunoassay/enzyme-linked immunofiltration assay with integrated negative control reaction. J. Agric. Food Chem. 1995, 43, 2548–2552. [Google Scholar]
- Collin, R.; Schneider, E.; Briggs, L.; Towers, N. Development of immunodiagnostic field tests for the detection of the mycotoxin, sporidesmin A. Food Agric. Immunol. 1998, 10, 91–104. [Google Scholar] [CrossRef]
- Sibanda, L.; De Saeger, S.; Van Peteghem, C.; Grabarkiewicz-Szczesna, J.; Tomczak, M. Detection of T-2 toxin in different cereals by flow-through enzyme immunoassay with a simultaneous internal reference. J. Agric. Food Chem. 2000, 48, 5864–5867. [Google Scholar] [PubMed]
- Mullett, W.; Lai, E.P.; Yeung, J.M. Immunoassay of fumonisins by a surface plasmon resonance biosensor. Anal. Biochem. 1998, 258, 161–167. [Google Scholar] [PubMed]
- Logrieco, A.; Arrigan, D.W.M.; Brengel-Pesce, K.; Siciliano, P.; Tothill, I.E. DNA arrays, electronic noses and tongues, biosensors and receptors for rapid detection of toxigenic fungi and mycotoxins: A review. Food Addit. Cont. 2005, 22, 335–344. [Google Scholar] [CrossRef]
- Tothill, I.E. Biosensors developments and potential applications in the agricultural diagnosis sector. Comp. Elect. Agric. 2001, 30, 205–218. [Google Scholar]
- Gaag, V.D.B.; Spath, S.; Dietrich, H.; Stigter, E.; Boonzaaijer, G.; van Osenbruggen, T.; Koopal, K. Biosensor and multiple mycotoxin analysis. Food Control 2003, 14, 251–254. [Google Scholar] [CrossRef]
- Daly, S.J.; Keating, G.J.; Dillon, P.P.; Manning, B.M.; O’Kennedy, R.; Lee, H.A.; Morgan, R.A. Development of surface plasmon resonance-based immunoassay for aflatoxin B1. J. Agric. Food Chem. 2000, 48, 5097–5104. [Google Scholar] [PubMed]
- Thompson, V.S.; Maragos, C.M. Fiber–optic Immunosensor for the detection of fumonisin B1. J. Agric. Food Chem. 1996, 44, 1041–1046. [Google Scholar]
- Tothill, I.E. On-line immunochemical assays for contaminant analysis. In Rapid and on-line Instrumentation for Food Quality Assurance; Tothill, I.E., Ed.; Woodhead Publishing Limited: Cambridge, UK, 2003; pp. 15–39. [Google Scholar]
- Salam, F.; Tothill, I.E. Detection of Salmonella typhimurium using an electrochemical immunosenor. Biosen. Bioelec. 2009, 24, 2630–2636. [Google Scholar] [CrossRef] [Green Version]
- Parker, O.P.; Tothill, I.E. Development of an electrochemical immunosensor for aflatoxin M1 in milk wit focus on matrix interference. Biosen. Bioelec. 2009, 24, 2452–2457. [Google Scholar]
- Crew, A.; Alford, C.; Cowell, D.C.C.; Hart, J.P. Development of a novel electrochemical immuno-assay using a screen printed electrode for the determination of secretory immunoglobulin a in human sweat. Electroch. Acta 2007, 52, 5232–5237. [Google Scholar]
- Butler, D.; Guilbault, G.G. Disposable amperometric immunosensor for the detection of 17-ß estradiol using screen-printed electrodes. Sens. Actuat. B 2006, 113, 692. [Google Scholar]
- Micheli, L.; Grecco, R.; Badea, M.; Moscone, D.; Palleschi, G. An electrochemical immunosensor for aflatoxin M1 determination in milk using screen-printed electrodes. Biosens. Bioelec. 2005, 21, 588–596. [Google Scholar] [Green Version]
- Studentsov, Y.Y.; Schiffman, M.; Strickler, H.D.; Ho, G.Y.F.; Susana Pang, Y.Y.; Schiller, J.; Herrero, R.; Burk, R.D. Enhanced enzyme-linked immunosorbent assay for detection of antibodies to virus-like particles of human papillomavirus. J. Clin. Microbil. 2002, 40, 1755–1760. [Google Scholar]
- Warwick, M.J. Chap 8: Standardisation of immunoassay. In Immunoassay, A Practical Guide; Brian, L., Ed.; Taylor & Francis Ltd.: London, UK, 1996; p. 160. [Google Scholar]
- Ammida, N.H.S.; Micheli, L.; Palleschi, G. Electrochemical immunosensor for determination of aflatoxin B1 in barley. Analy. Chim. Acta 2004, 50, 159–164. [Google Scholar]
- Alarcon, S.H.; Palleschi, G.; Compagnone, D.; Pascale, M.; Visconti, A.; Barna-Vetro, I. Monoclonal antibody based electrochemical immunosensor for the determination of ochratoxin A in wheat. Talanta 2006, 69, 1031–1037. [Google Scholar] [PubMed]
- Volpe, G.; Compagnone, D.; Draisci, R.; Palleschi, G. 3,3,5,5-Tetramethylbenzidine as electrochemical substrate for horseradish peroxidase based enzyme immunoassays. A comparative study. Analyst 1998, 123, 1303–1307. [Google Scholar] [CrossRef]
- Fu, X.H. Electrochemical measurement of DNA hybridization using nanosilver as label and horseradish peroxidase as enhancer. Bioprocess Biosyst. Eng. 2008, 31, 69–73. [Google Scholar]
- Muscarella, M.; Margo, L.S.; Nardiello, D.; Palermo, C.; Centonze, D. Development of a new analytical method for the determination of fumonisin B1 and B2 in food products based on high performance liquid chromatography and fluorimetric detection with post-column derivatization. J. Chrom. A. 2008, 1203, 88–93. [Google Scholar]
- Noh, M.F.M.; Tothill, I.E. Development and characterisation of disposable gold electrodes, and their use for lead (II) analysis. Anal. Bioanal. Chem. 2006, 386, 2095–2210. [Google Scholar] [CrossRef] [PubMed]
- Anna, Y.K.; Shim, W.B.; Yang, Z.Y.; Eremin, S.A.; Chung, D.H. Direct competitive ELISA based on a monoclonal antibody for detection of aflatoxin B1. Stabilization of ELISA kit components and application to grain samples. Anal. Bioanal. Chem. 2006, 384, 286–294. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abdul Kadir, M.K.; Tothill, I.E. Development of an Electrochemical Immunosensor for Fumonisins Detection in Foods. Toxins 2010, 2, 382-398. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2040382
Abdul Kadir MK, Tothill IE. Development of an Electrochemical Immunosensor for Fumonisins Detection in Foods. Toxins. 2010; 2(4):382-398. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2040382
Chicago/Turabian StyleAbdul Kadir, Mohamad Kamal, and Ibtisam E. Tothill. 2010. "Development of an Electrochemical Immunosensor for Fumonisins Detection in Foods" Toxins 2, no. 4: 382-398. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins2040382