Uremic Toxins Enhance Statin-Induced Cytotoxicity in Differentiated Human Rhabdomyosarcoma Cells

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
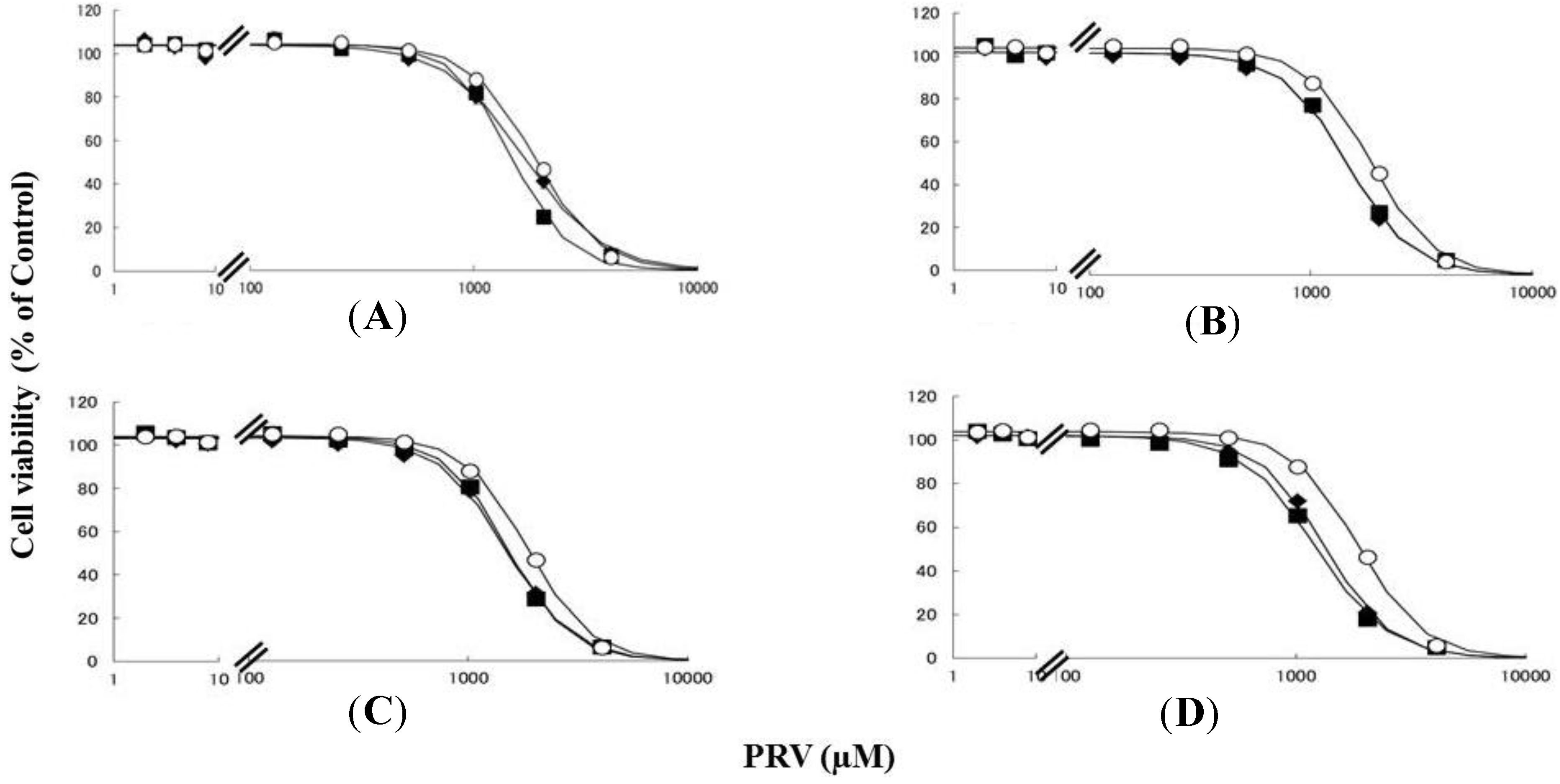
2.1. Effects of Pre-Treatment with Uremic Toxin (CMPF, Hippuric Acid, Indole-3-Acetic Acid, or 3-Indolxyl Sulfate) on Statin-Induced Cytotoxicity in Differentiated RD Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uremic toxins | LC50 Values (95% Confidence Interval) | ||
|---|---|---|---|
| PRV (µM) | SIM (µM) | ||
| Untreated | 1865 (1819–1919) | 6.30 (6.05–6.54) | |
| CMPF | 2 µM | 1675 (1570–1779) * | 5.73 (5.17–6.29) * |
| 400 µM | 1438 (1398–1549) *,† | 5.05 (4.61–5.50) * | |
| Hippuric acid | 180 µM | 1463 (1420–1506) * | 5.79 (5.46–6.11) |
| 400 µM | 1463 (1419–1506) * | 5.37 (5.06–5.68) * | |
| Indole-3-acetic acid | 3 µM | 1484 (1438–1529) * | 5.84 (5.40–6.28) |
| 20 µM | 1513 (1464–1583) * | 5.70 (5.36–6.04) * | |
| 3-indoxyl sulfate | 20 µM | 1342 (1303–1381) * | 5.49 (5.18–5.79) * |
| 200 µM | 1225 (1188–1261) *,† | 4.57 (4.34–4.80) *,† | |
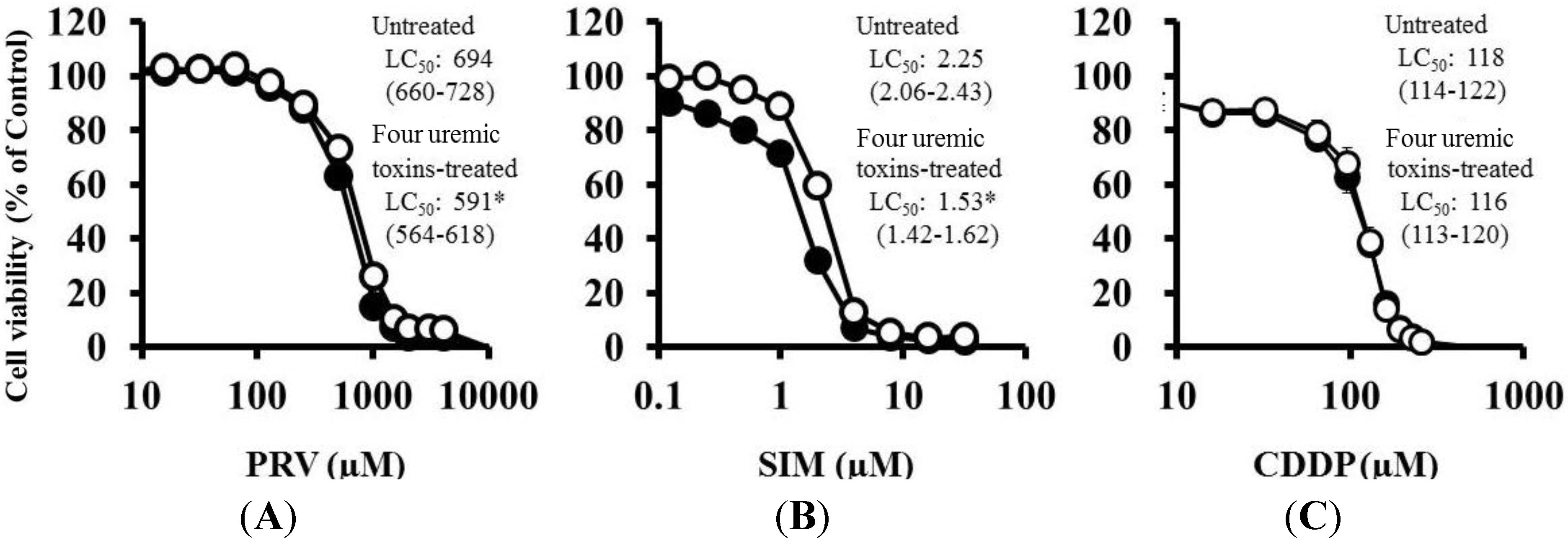
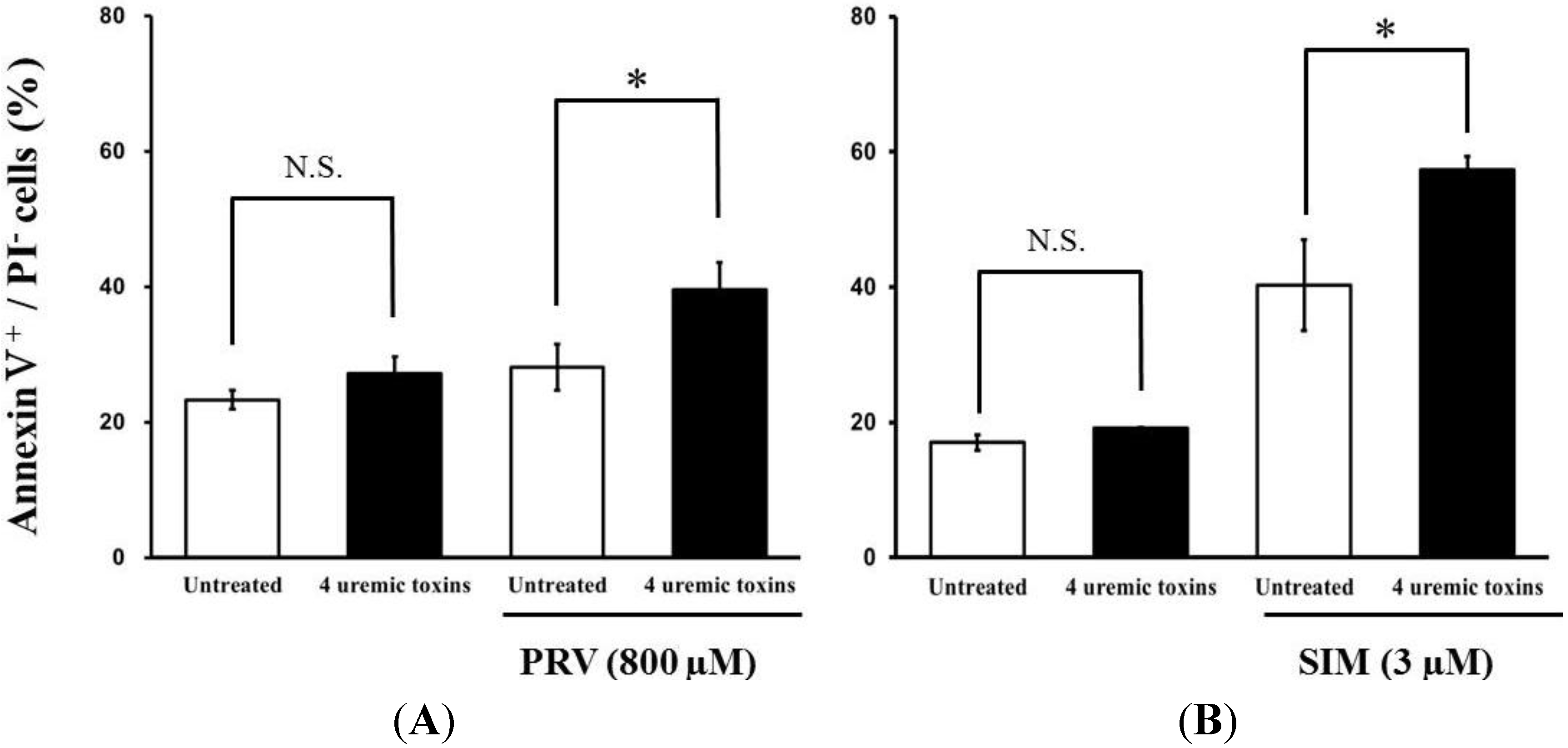
2.2. Effects of Pre-treatment with Uremic Toxins on Statin-Induced Cytotoxicity and Apoptosis in Differentiated RD Cells


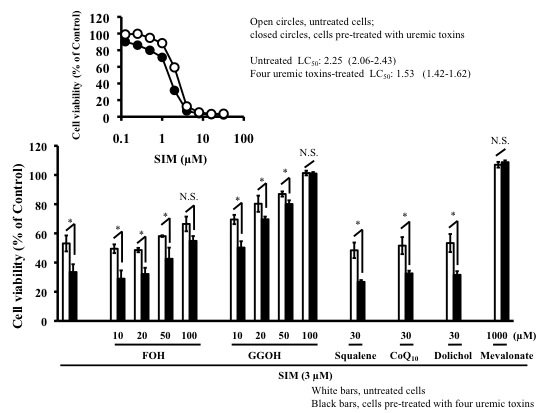
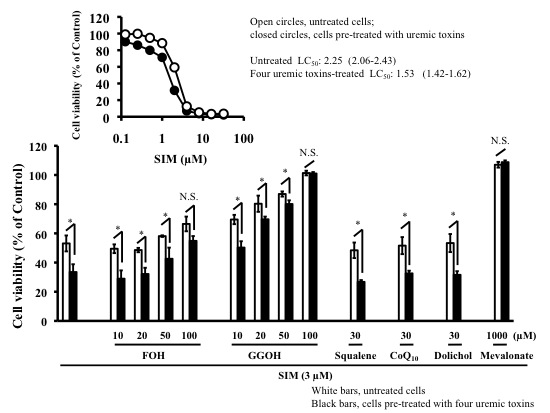
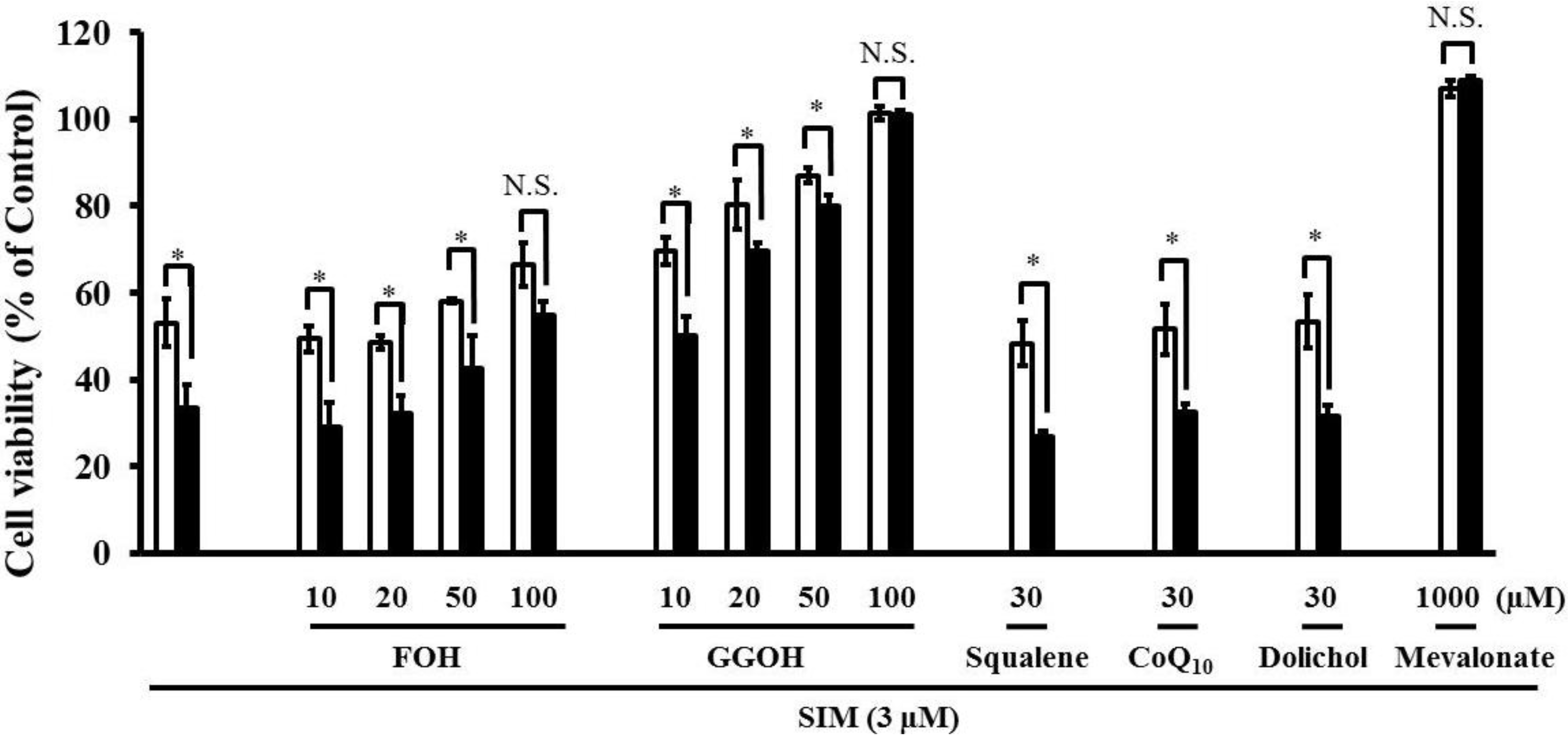
2.3. Effects of Mevalonate Pathway-Related Compounds on Statin-Induced Apoptosis in Differentiated RD Cells Treated with Four Uremic Toxins

| LC50 Values (95% Confidence Interval) | ||||
|---|---|---|---|---|
| PRV (µM) | PRV (µM) + Mevalonate (µM) | SIM (µM) | SIM (µM) + Mevalonate (µM) | |
| Untreated | 898 (857–940) | N.C. | 2.23 (2.15–2.31) | 105 (103–106) |
| Four uremic toxins | 814 (786–842) * | N.C. | 1.97 (1.89–2.04) * | 103 (102–104) |

3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.3. Evaluation of Cytotoxicity
3.4. Assessment of Apoptosis
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, H.J.; Kim, E.K.; Kwon, S.U.; Kim, J.S.; Kang, D.W. Effect of statin on progression of symptomatic intracranial atherosclerosis. Can. J. Neurol. Sci. 2012, 39, 801–806. [Google Scholar] [PubMed]
- Shah, S.; Paparello, J.; Danesh, F.R. Effects of statin therapy on the progression of chronic kidney disease. Adv. Chronic Kidney Dis. 2005, 12, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Cobbe, S.M.; Ford, I.; Isles, C.G.; Lorimer, A.R.; MacFarlane, P.W.; McKillop, J.H.; Packard, C.J. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med. 1995, 20, 1301–1307. [Google Scholar] [CrossRef]
- Schech, S.; Graham, D.; Staffa, J.; Andrade, S.E.; la Grenade, L.; Burgess, M.; Blough, D.; Stergachis, A.; Chan, K.A.; Platt, R.; et al. Risk factors for statin-associated rhabdomyolysis. Pharmacoepidemiol. Drug Saf. 2007, 16, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Gehr, T.W.; Sica, D.A.; Slugg, P.H.; Hammett, J.L.; Raymond, R.; Ford, N.F. The pharmacokinetics of pravastatin in patients on chronic hemodialysis. Eur. J. Clin. Pharmacol. 1997, 53, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Lins, R.L.; Matthys, K.E.; Verpooten, G.A.; Peeters, P.C.; Dratwa, M.; Stolear, J.C.; Lameire, N.H. Pharmacokinetics of atorvastatin and its metabolites after single and multiple dosing in hypercholesterolaemic haemodialysis patients. Nephrol. Dial. Transplant. 2003, 18, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Quérin, S.; Lambert, R.; Cusson, J.R.; Grégoire, S.; Vickers, S.; Stubbs, R.J.; Sweany, A.E.; Larochelle, P. Single-dose pharmacokinetics of 14C-lovastatin in chronic renal failure. Clin. Pharmacol. Ther. 1991, 50, 437–441. [Google Scholar]
- Singhvi, S.M.; Pan, H.Y.; Morrison, R.A.; Willard, D.A. Disposition of pravastatin sodium, a tissue-selective HMG-CoA reductase inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 1990, 29, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Bogsrud, M.P.; Molden, E.; Asberg, A.; Mohebi, B.U.; Ose, L.; Retterstøl, K. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin. Pharmacol. Ther. 2006, 79, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin. Pharmacol. Ther. 2006, 80, 356–366. [Google Scholar] [PubMed]
- Pasanen, M.K.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genomics 2006, 16, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, M.K.; Fredrikson, H.; Neuvonen, P.J.; Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2007, 82, 726–733. [Google Scholar] [PubMed]
- Voora, D.; Shah, S.H.; Spasojevic, I.; Ali, S.; Reed, C.R.; Salisbury, B.A.; Ginsburg, G.S. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 2009, 54, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- De Melo, M.P.; de Lima, T.M.; Pithon-Curi, T.C.; Curi, R. The mechanism of indole acetic acid cytotoxicity. Toxicol. Lett. 2004, 148, 103–111. [Google Scholar]
- Miyamoto, Y.; Iwao, Y.; Mera, K.; Watanabe, H.; Kadowaki, D.; Ishima, Y.; Chuang, V.T.; Sato, K.; Otagiri, M.; Maruyama, T. A uremic toxin, 3-carboxy-4-methyl-5-propyl-2-furanpropionate induces cell damage to proximal tubular cells via the generation of a radical intermediate. Biochem. Pharmacol. 2012, 84, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Niwa, T. Indoxyl sulfate promotes proliferation of human aortic smooth muscle cells by inducing oxidative stress. J. Ren. Nutr. 2009, 19, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Spustová, V.; Dzúrik, R.; Geryková, M. Hippurate participation in the inhibition of glucose utilization in renal failure. Czechosolv. Med. 1987, 10, 79–89. [Google Scholar]
- Itagaki, M.; Takaguri, A.; Kano, S.; Kaneta, S.; Ichihara, K.; Satoh, K. Possible mechanisms underlying statin-induced skeletal muscle toxicity in L6 fibroblasts and in rats. J. Pharmacol. Sci. 2009, 109, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Suzuki, M.; Aoki, T.; Morikawa, S.; Maejima, T.; Sato, F.; Sawanobori, K.; Kitahara, M.; Kodama, T.; Saito, Y.; et al. Influence of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on ubiquinone levels in rat skeletal muscle and heart: Relationship to cytotoxicity and inhibitory activity for cholesterol synthesis in human skeletal muscle cells. J. Atheroscler. Thromb. 2006, 13, 295–307. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Y.; Tang, R.; Chen, Y.; Li, Q.; Gong, J.; Huang, A.; Varghese, Z.; Moorhead, J.F.; Ruan, X.Z.; et al. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. J. Gastroenterol. Hepatol. 2011, 26, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Chisaki, I.; Narumi, K.; Hidaka, K.; Kagawa, T.; Itagaki, S.; Hirano, T.; Iseki, K. Association between risk of myopathy and cholesterol-lowering effect: A comparison of all statins. Life Sci. 2008, 82, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Tanida, S.; Mizoshita, T.; Ozeki, K.; Tsukamoto, H.; Kamiya, T.; Kataoka, H.; Sakamuro, D.; Joh, T. Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: Potential of BIN1 to act as a potent predictor of cisplatin sensitivity in gastric cancer treatment. Int. J. Surg. Oncol. 2012, 2012. [Google Scholar] [CrossRef]
- Zhong, W.B.; Liang, Y.C.; Wang, C.Y.; Chang, T.C.; Lee, W.S. Lovastatin suppresses invasiveness of anaplastic thyroid cancer cells by inhibiting Rho geranylgeranylation and RhoA/ROCK signaling. Endocr. Relat. Cancer 2005, 12, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.E.; Zhang, X.; Bleicher, K.B.; Dysart, G.; Loughlin, A.F.; Schaefer, W.H.; Umbenhauer, D.R. Statins induce apoptosis in rat and human myotube cultures by inhibiting protein geranylgeranylation but not ubiquinone. Toxicol. Appl. Pharmacol. 2004, 200, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the pharmacokinetics and redox properties of protein-bound uremic toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, Q.; Wang, L.; Chen, Y.; Wang, L.; Zhang, W. Interleukin-1β enhances the intracellular accumulation of cholesterol by up-regulating the expression of low-density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase in podocytes. Mol. Cell. Biochem. 2011, 346, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chida, A.S.; Rahman, I. Redox modifications of protein-thiols: Emerging roles in cell signaling. Biochem. Pharmacol. 2006, 71, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Backman, J.T.; Kyrklund, C.; Kivistö, K.T.; Wang, J.S.; Neuvonen, P.J. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin. Pharmacol. Ther. 2000, 68, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Lilja, J.J.; Kivistö, K.T.; Neuvonen, P.J. Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin. Pharmacol. Ther. 1999, 66, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kaido, F.; Kagawa, T.; Itagaki, S.; Hirano, T.; Iseki, K. Preventive effects of bicarbonate on cerivastatin-induced apopotosis. Int. J. Pharm. 2007, 341, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Pazzagli, C.; Born, T.L.; Bertolaet, B.L.; Knudsen, K.E.; Arden, K.C.; Henry, R.R.; Feramisco, J.R. Elevated cyclins and cyclin-dependent kinase activity in the rhabdomyosarcoma cell line RD. Cancer Res. 1998, 58, 2042–2049. [Google Scholar]
- Nishimoto, T.; Tozawa, R.; Amano, Y.; Wada, T.; Imura, Y.; Sugiyama, Y. Comparing myotoxic effects of squalene synthase inhibitor, T-91485, and 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors in human myocytes. Biochem. Pharmacol. 2003, 66, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Liebich, H.M.; Bubeck, J.I.; Picker, A.; Wahl, G.; Scheiter, A. Hippuric acid and 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid in serum and urine. Analytical approaches and clinical relevance in kidney diseases. J. Chromatogr. 1990, 500, 615–627. [Google Scholar]
- Sakai, T.; Maruyama, T.; Imamura, H.; Shimada, H.; Otagiri, M. Mechanism of stereoselective serum binding of ketoprofen after hemodialysis. J. Pharmacol. Exp. Ther. 1996, 278, 786–792. [Google Scholar] [PubMed]
- Tsujimoto, M.; Kinoshita, Y.; Hirata, S.; Otagiri, M.; Ohtani, H.; Sawada, Y. Effects of uremic serum and uremic toxins on hepatic uptake of digoxin. Ther. Drug Monit. 2008, 30, 576–582. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Uchiyama, H.; Tsujimoto, M.; Shinmoto, T.; Ogino, H.; Oda, T.; Yoshida, T.; Furukubo, T.; Izumi, S.; Yamakawa, T.; Tachiki, H.; et al. Uremic Toxins Enhance Statin-Induced Cytotoxicity in Differentiated Human Rhabdomyosarcoma Cells. Toxins 2014, 6, 2612-2625. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins6092612
Uchiyama H, Tsujimoto M, Shinmoto T, Ogino H, Oda T, Yoshida T, Furukubo T, Izumi S, Yamakawa T, Tachiki H, et al. Uremic Toxins Enhance Statin-Induced Cytotoxicity in Differentiated Human Rhabdomyosarcoma Cells. Toxins. 2014; 6(9):2612-2625. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins6092612
Chicago/Turabian StyleUchiyama, Hitoshi, Masayuki Tsujimoto, Tadakazu Shinmoto, Hitomi Ogino, Tomoko Oda, Takuya Yoshida, Taku Furukubo, Satoshi Izumi, Tomoyuki Yamakawa, Hidehisa Tachiki, and et al. 2014. "Uremic Toxins Enhance Statin-Induced Cytotoxicity in Differentiated Human Rhabdomyosarcoma Cells" Toxins 6, no. 9: 2612-2625. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins6092612