
Pharmacological Alternatives for the Treatment of Neurodegenerative Disorders: Wasp and Bee Venoms and Their Components as New Neuroactive Tools

Abstract
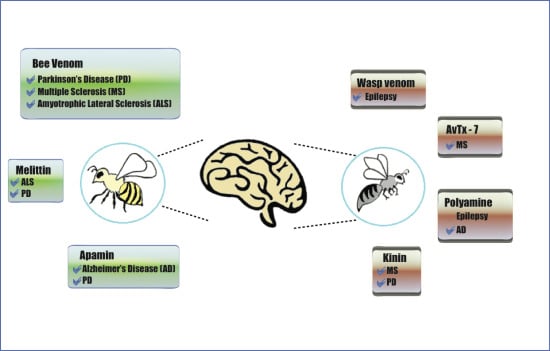
:
1. Introduction
2. General Profile of the Main Neurodegenerative Diseases

2.1. Alzheimer’s Disease and Other Dementias
2.2. Parkinson’s Disease
2.3. Epilepsy
2.4. Multiple Sclerosis
2.5. Amyotrophic Lateral Sclerosis
3. Venoms and Toxins from Wasps and Bees to Combat Neurodegenerative Disorders
3.1. Bee Venom

{kind=link}
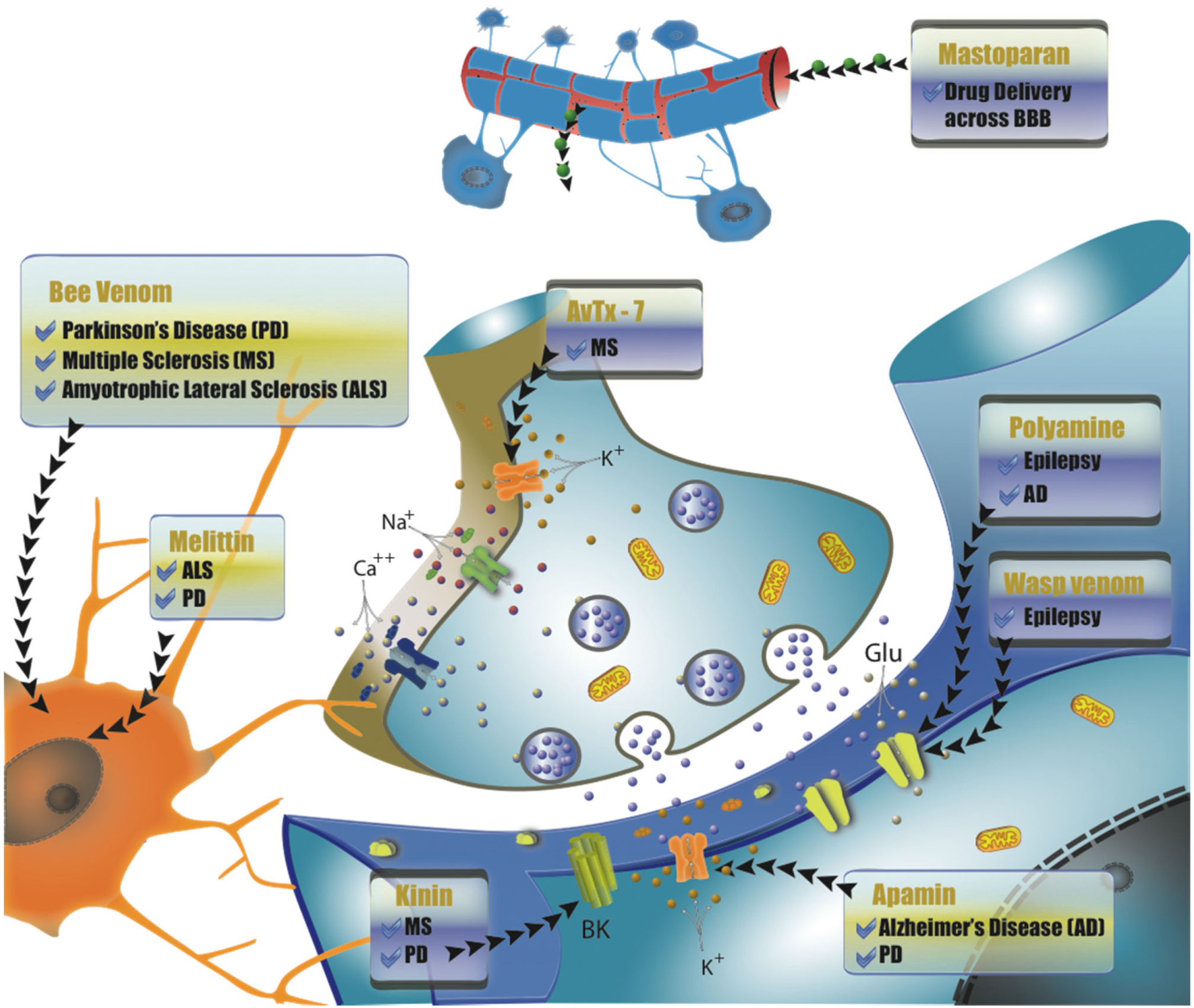{kind=link}
{kind=link}
| Venom or Compound | Neurological Disease | Model Tested | Administration via | Dose | Reference |
|---|---|---|---|---|---|
| Bee venom | Parkinson’s Disease | 1-methyl-4-phenyl-1,2,4,5-tetrahydropyridine (MPTP) in mice | s.c. acupuncture (point GB34) | 0.02 mL bee venom (1:2000 w/v) | [81] |
| once every 3 days for 2 weeks | |||||
| Bee venom | Parkinson’s Disease | MPTP in mice | s.c. acupuncture (bilateral point ST36) | A single injection (0.6 mg/kg) | [82] |
| Bee venom | Parkinson’s Disease | MPTP/probenecid in mice | i.p. | Two injections 3.5 days apart for 5 weeks | [83] |
| Low—12 µg/kg/BW | |||||
| High—120 µg/kg/BW | |||||
| Bee venom | Parkinson’s Disease | MPTP in mice | i.p. | one i.p. injection | [84] |
| BV (1 mg/kg) every day for 6 days | |||||
| Bee venom | Parkinson’s Disease | Rotenone-induced oxidative stress and apoptosis | s.c. acupuncture (point GB34) | 0.02 mL bee venom (1:2000 w/v) | [85] |
| once every 3 days for 2 weeks | |||||
| Bee venom | Multiple Sclerosis | Experimental allergic encephalomyelitis model in rats | - | 2 mg/kg or 5 mg/kg | [86] |
| Bee venom | Amyotrophic Lateral Sclerosis | hSOD1G93A transgenic mice | s.c. acupuncture (bilateral point ST36) | 0.1 µg/g—3 times/week for 2 weeks | [87] |
| Bee venom | Amyotrophic Lateral Sclerosis | hSOD1G93A transgenic mice | s.c. acupuncture (bilateral point ST36) i.p. | 0.1 µg/g—3 times/week for 2 weeks | [88] |
| Apamin | Parkinson’s Disease | MPTP/probenecid mice | i.p. | Two injections 3.5 days apart for 5 weeks | [83] |
| Low—0.5 µg/kg/BW | |||||
| High—1.0 µg/kg/BW | |||||
| Melittin | Amyotrophic Lateral Sclerosis | hSOD1G93A transgenic mice | s.c. acupuncture (bilateral point ST36) | 0.1 µg/g twice a week | [89] |
3.2. Wasp Venom
| Venom or Compound | Neurological Disease | Model Tested | Route of Administration | Dose | Reference |
|---|---|---|---|---|---|
| Denatured venom—P. ignobilis | Epilepsy | Acute seizures model induced by chemoconvulsants in rats | i.c.v. | 400 μg/animal | [104] |
| Denatured venom—P. occidentalis | Epilepsy | Acute seizures model induced by chemoconvulsants in rats | i.c.v. | 120, 240 and 300 μg/animal | [105] |
| Low molecular weight compounds—P. occidentalis | Epilepsy | Acute seizures model induced by PTZ | i.c.v. | 70, 210 and 350 μg/animal | [106] |
| Bradykinin | Stroke | Transient forebrain ischemia in rats | i.p. | 150 μg/kg 48 h after ischemia | [107] |
| Bradykinin | Stroke | Transient forebrain ischemia in rats | i.p. | 150 μg/kg 48 h after ischemia | [108] |
4. Compounds Isolated from Wasp and Bee Venom for the Treatment of Neurodegenerative Diseases
4.1. Peptides from Bee Venom as Therapeutic Sources
4.1.1. Melittin

4.1.2. Apamin
4.2. Wasp Venom Peptides as Therapeutic Sources
4.2.1. Pompilidotoxins
4.2.2. AvTx-7
4.2.3. Mastoparan
4.2.4. Wasp Kinin
4.3. Polyamine Toxins as Therapeutic Sources
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bogdanov, S. Bee venom: Composition, health, medicine: A review. Peptides 2015, 1, 1–20. [Google Scholar]
- Pemberton, R.W. Insects and other arthropods used as drugs in Korean traditional medicine. J. Ethnopharmacol. 1999, 65, 207–216. [Google Scholar] [CrossRef]
- Adewole, A.M.; Ileke, K.D.; Oluyede, P.O. Perception and knowledge of bee venom therapy as an alternative treatment for common ailments in southwestern Nigeria. FUTA J. Res. Sci. 2013, 9, 235–240. [Google Scholar]
- Ali, M.A. Studies on bee venom and its medical uses. Int. J. Adv. Res. Technol. 2012, 1, 69–83. [Google Scholar]
- Santos, L.D.; Pieroni, M.; Menegasso, A.R.S.; Pinto, J.R.A.S.; Palma, M.S. A new scenario of bioprospecting of Hymenoptera venoms through proteomic approach. J. Venom. Anim. Toxins Incl. Trop. Dis. 2011, 17, 364–377. [Google Scholar]
- Mortari, M.R.; Cunha, A.O.S.; Ferreira, L.B.; dos Santos, W.F. Neurotoxins from invertebrates as anticonvulsants: From basic research to therapeutic application. Pharmacol. Ther. 2007, 114, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Mortari, M.R.; Cunha, A.O.S. New perspectives in drug discovery using neuroactive molecules from the venom of Arthropods. In An Integrated View of the Molecular Recognition and Toxinology—From Analytical Procedures to Biomedical Applications; Radis-Baptista, G., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Di Filippo, M.; Ghiglieri, V.; Tambasco, N.; Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s Disease: Filling the bench-to-bedside gap. Lancet Neurol. 2010, 9, 1106–1117. [Google Scholar] [CrossRef]
- Hung, A.Y.; Schwarzschild, M.A. Treatment of Parkinson’s Disease: What’s in the non-dopaminergic pipeline? Neurotherapeutics 2014, 11, 34–46. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Neurological Disorders: Public Health Challengers. 2006. Available online: http://www.who.int/mental_health/neurology/neurological_disorders_report_web.pdf (accessed on 10 March 2015).
- Escoubas, P.; Quinton, L.; Nicholson, G.M. Venomics: Unravelling the complexity of animal venoms with mass spectrometry. J. Mass Spectrom. 2008, 43, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Ménez, A.; Stöcklin, R.; Mebs, D. ‘Venomics’ or: The venomous systems genome project. Toxicon 2006, 47, 255–259. [Google Scholar] [CrossRef] [PubMed]
- De Boer, H.M.; Mula, M.; Sander, J.W. The global burden and stigma of epilepsy. Epilepsy Behav. 2008, 12, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, A. Stigma, epilepsy, and quality of life. Epilepsy Behav. 2002, 3, 10–20. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Prvulovic, D.; Teipel, S.; Jessen, F.; Luckhaus, C.; Frölich, L.; Riepe, M.W.; Dodel, R.; Leyhe, T.; Bertram, L.; et al. The future of Alzheimer’s Disease: The next 10 years. Prog. Neurobiol. 2011, 95, 718–728. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia: A Public Health Priority. 2012. Available online: http://whqlibdoc.who.int/publications/2012/9789241564458_eng.pdf (accessed on 10 March 2015).
- Wimo, A.; Winblad, B.; Aguero-Torres, H.; von Strauss, E. The magnitude of dementia occurrence in the world. Alzheimer Dis. Assoc. Disord. 2003, 17, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Davey, D.A. Alzheimer’s Disease and vascular dementia: One potentially preventable and modifiable disease? Part II: Management, prevention and future perspective. Neurodegener. Dis. Manag. 2014, 4, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ekavali, A.S. A review on Alzheimer’s Disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Perry, G. Pathogenesis and disease-modifying therapy in Alzheimer’s Disease: The flat line of progress. Arch. Med. Res. 2012, 43, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Frölich, L. The cholinergic pathology in Alzheimer’s Disease—Discrepancies between clinical and pathophysiological findings. J. Neural. Transm. 2002, 109, 1003–1014. [Google Scholar] [PubMed]
- Schneider, L.S.; Dagerman, K.S.; Higgins, J.P.; McShane, R. lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch. Neurol. 2011, 68, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Obeso, J.A. The significance of defining preclinical or prodromal Parkinson’s Disease. Mov. Disord. 2012, 27, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Moon, C.S.; Khogali, A.; Haidous, A.; Chabenne, A.; Ojo, C.; Jelebinkov, M.; Kurdi, Y.; Ebadi, M. Biomarkers in Parkinson’s disease (recent update). Neurochem. Int. 2013, 63, 201–229. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s Disease as a result of aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s Disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singh, S.; Sharma, V.; Singh, V.P.; Deshmukh, R. Neurobiology of l-DOPA induced dyskinesia and the novel therapeutic strategies. Biomed. Pharmacother. 2015, 70, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Dahiya, N. Management of Parkinson’s Disease: Current and future pharmacotherapy. Eur. J. Pharmacol. 2015, 750, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J.J. Epileptic seizures and epilepsy: Definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; Frech, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-resistant epilepsy. N. Engl. J. Med. 2011, 365, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Schachter, S.C. Drug treatment of epilepsy in adults. BMJ 2014, 348, g254. [Google Scholar] [CrossRef] [PubMed]
- Simonato, M.; Löscher, W.; Cole, A.J.; Dudek, F.E.; Engel, J., Jr.; Kaminski, R.M.; Loeb, J.A.; Scharfman, H.; Staley, K.J.; Velisek, L.; et al. Finding a better drug for epilepsy: Preclinical screening strategies and experimental trial design. Epilepsia 2012, 53, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.; Harding, K.E.; Clarkson, H.; Pickersgill, T.P.; Wardle, M.; Robertson, N.P. Demographic and clinical factors associated with changes in employment in multiple sclerosis. Mult. Scler. 2013, 19, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Pawate, S.; Bagnato, F. Newer agents in the treatment of multiple sclerosis. Neurologist 2015, 19, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Sand, I.K. Classification, diagnosis, and differential diagnosis of multiple sclerosis. Curr. Opin. Neurol. 2015, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- National Multiple Sclerosis Society. Available online: http://www.nationalmssociety.org/What-is-MS/Who-Gets-MS (accessed on 10 April 2015).
- Lublin, F.D. New multiple sclerosis phenotypic classification. Eur. Neurol. 2014, 72, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Fox, R.J. Progressive multiple sclerosis. Curr. Opin. Neurol. 2015, 28, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rovaris, M.; Confavreux, C.; Furlan, R.; Kappos, L.; Comi, G.; Filippi, M. Secondary progressive multiple sclerosis: Current knowledge and future challenges. Lancet Neurol. 2006, 5, 343–354. [Google Scholar] [CrossRef]
- Lassman, H. Multiple sclerosis: Is there neurodegeneration independent from inflammation? J. Neurol. Sci. 2007, 259, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Hyland, M.; Cohen, J.A. Multiple sclerosis: New insights in pathogenesis and novel therapeutics. Annu. Rev. Med. 2012, 63, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Giordana, M.T.; Ferrero, P.; Grifoni, S.; Pellerino, A.; Naldi, A.; Montuschi, A. Dementia and cognitive impairment in amyotrophic lateral sclerosis: A review. Neurol. Sci. 2011, 32, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, C.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-Derived TGF-β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep. 2015, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Freer, C.; Hylton, T.; Jordan, H.M.; Kaye, W.E.; Singh, S.; Huang, Y. Results of Florida’s Amyotrophic Lateral Sclerosis Surveillance Project, 2009–2011. BMJ Open 2015, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jelsone-Swain, L.; Persad, C.; Burkard, D.; Welsh, R.C. Action Processing and Mirror Neuron Function in Patients with Amyotrophic Lateral Sclerosis: An fMRI Study. PLoS ONE 2015, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Gelati, M.; Profico, D.C.; Sgaravizzi, G.; Pensi, M.P.; Muzi, G.; Ricciolini, C.; Nodari, L.R.; Carletti, S.; Giorgi, C.; et al. Human neural stem cell transplantation in ALS: Initial results from a phase I trial. J. Transl. Med. 2015, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Lunn, J.S.; Sakowski, S.A.; Feldman, E.L. Concise review: Stem cell therapies for amyotrophic lateral sclerosis: Recent advances and prospects for the future. Stem Cells 2014, 32, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Achi, E.Y.; Rudnicki, S.A. ALS and Frontotemporal Dysfunction: A Review. Neurol. Res. Int. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.I.; Park, A.; Kim, H.J.; Oh, K.W.; Choi, H.; Kwon, M.J.; Ki, C.S.; Kim, H.T.; Kim, S.H. Spectrum of cognitive impairment in Korean ALS patients without known genetic mutations. PLoS ONE 2014, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, A.; Iazzolino, B.; Calvo, A.; Moglia, C.; Lopiano, L.; Restagno, G.; Brunetti, M.; Ossola, I.; lo Presti, A.; Cammarosano, S.; et al. Cognitive correlates in amyotrophic lateral sclerosis: A population-based study in Italy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, C.; Mitrecic, D.; Falnikar, A.; Lepore, A.C. Transplantation of stem cell-derived astrocytes for the treatment of amyotrophic lateral sclerosis and spinal cord injury. World J. Stem Cells 2015, 7, 380–398. [Google Scholar] [CrossRef] [PubMed]
- Borasio, G.D.; Miller, R.G. Clinical characteristics and management of ALS. Semin. Neurol. 2001, 21, 155–166. [Google Scholar] [CrossRef] [PubMed]
- De Lima, P.R.; Brochetto-Braga, M.R. Hymenoptera venom review focusing on Apis mellifera. J. Venom. Anim. Toxins incl. Trop. Dis. 2003. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeon, B.S. Is acupuncture efficacious therapy in Parkinson’s Disease? J. Neurol. Sci. 2014, 341, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ezzo, J.; Hadhazy, V.; Birch, S.; Lao, L.; Kaplan, G.; Hochberg, M.; Berman, B. Acupuncture for osteoarthritis of the knee: A systematic review. Arthritis Rheum. 2001, 44, 819–825. [Google Scholar] [CrossRef]
- Lee, M.S.; Pittler, M.H.; Shin, B.C.; Kong, J.C.; Ernst, E. Bee venom acupuncture for musculoskeletal pain: A review. J. Pain 2008, 9, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Park, H.J.; Chae, Y.; Lim, S. An overview of bee venom acupuncture in the treatment of arthritis. Evid. Based Complement. Alternat. Med. 2005, 2, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Castro, H.J.; Mendez-Lnocenio, J.I.; Omidvar, B.; Omidvar, J.; Santilli, J.; Nielsen, H.S., Jr.; Pavot, A.P.; Richert, J.R.; Bellanti, J.A. A phase I study of the safety of honeybee venom extract as a possible treatment for patients with progressive forms of multiple sclerosis. Allergy Asthma Proc. 2005, 26, 470–476. [Google Scholar] [PubMed]
- Kwon, Y.B.; Lee, J.H.; Han, H.J.; Mar, W.C.; Beitz, A.J.; Lee, H.J. Bee venom injection into an acupunture point reduces arthritis associated edema and nociceptive responses. Pain 2001, 90, 271–280. [Google Scholar] [CrossRef]
- Kwon, Y.B.; Kim, H.W.; Ham, T.W.; Yoon, S.Y.; Roh, D.H.; Jan, H.J.; Beitz, H.J.; Yang, I.S.; Lee, J.H. The anti-inflammatory effect of bee venom stimulation in a mouse air pouch model is mediated by adrenal medullary activity. J. Neuroendocrinol. 2003, 15, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Pak, S.C.; Choi, S.H. The effect of whole bee venom on arthritis. Am. J. Chin. Med. 2002, 30, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.J.; Kim, K.S.; Kim, M.J.; Chang, Y.C.; Lee, S.D.; Kim, M.S.; Kim, C.H. Effects of bee venom on protease activities and free radical damages in synovial fluid from type II collagen-induced rheumatoid arthritis rats. Toxicol. In Vitro 2006, 20, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Park, S.Y.; Lee, K.J.; Heo, M.S.; Kim, K.C.; Kim, M.O.; Lee, J.D.; Choi, Y.H.; Kim, G.Y. Bee venom and melittin reduce proinflammatory mediators in lipopolysaccharide-stimulated BV2 microglia. Int. Immunopharmacol. 2007, 7, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Mahomoodally, M.F.; Bhugun, V.; Chutterdharry, G. Complementary and alternative medicines use against neurodegenerative diseases. Adv. Pharmacol. Pharm. 2013, 1, 103–123. [Google Scholar]
- Kim, S.U.; Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Shim, S.; Rhee, H.; Park, H.; Jung, W.; Moon, S.; Park, J.; Ko, C.; Cho, K.; Park, S. Effectiveness of acupuncture and bee venom acupuncture in idiopathic Parkinson’s Disease. Pakinsonism Relat. Disord. 2012, 18, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Mirshafiey, A. Venom therapy in multiple sclerosis. Neuropharmacology 2007, 53, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Doo, A.R.; Kim, S.T.; Kim, S.N.; Moon, W.; Yin, C.S.; Chae, Y.; Park, H.J. Neuroprotective effects of bee venom pharmaceutical acupuncture in acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s Disease. Neurol. Res. 2010, 32, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, J.; Kim, K.; Lee, W.; Kim, K.; Park, K. Melittin inhibits atherosclerosis in LPS/High-Fat treated mice through atheroprotective actions. J. Atheroscler. Thromb. 2011, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Fisher, D.; Noelker, C.; Vulinovic, F.; Grünewald, A.; Chevarin, C.; Klein, C.; Oertel, W.H.; Hirsch, E.C.; Michel, P.P.; Hartmann, A. Bee venom and its component Apamin as neuroprotetive agents in Parkinson disease mouse model. PLoS ONE 2013, 8, e61700. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Kim, H.; Lee, G.; Park, S.; Kim, H.; Bae, H. Neuro-protective effects of bee venom by suppression of neuroinflammatory responses in a mouse model of Parkinson’s Disease: Role of regulatory T cells. Brain Behav. Immun. 2012, 26, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Khalil, W.K.; Assaf, N.; ElShebiney, S.A.; Salem, N.A. Neuroprotective effects of bee venom acupuncture therapy against rotenone-induced oxidative stress and apoptosis. Neurochem. Int. 2014, 80, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Ahmadi, F.; Parivar, K.; Nabiuni, M.; Haghighi, S.; Imani, S.; Afrouzi, H. Effect of honey bee venom on lewis rats with experimental allergic encephalomyelitis, a model for multiple sclerosis. Iran. J. Pharm. Res. 2012, 11, 671–678. [Google Scholar] [PubMed]
- Yang, E.J.; Jiang, J.H.; Lee, S.M.; Yang, S.C.; Hwang, H.S.; Lee, M.S.; Choi, S.M. Bee venom attenuates neuroinflammatory events and extends survival in amyotrophic lateral sclerosis models. J. Neuroinflammation 2010, 15, 7–69. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Choi, S.M.; Yang, E.J. The effects of bee venom acupuncture on the central nervous system and muscle in an animal hSOD1G93A mutant. Toxins 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; Kim, J.M.; Park, K.K.; Chang, Y.C.; Pak, S.C. Neuroprotective effects of Melittin on hydrogen peroxide-induced apoptotic cell death in neuroblastoma SH-SY5Y cells. BMC Complement. Altern. Med. 2014, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Ham, T.W.; Yoon, S.Y.; Yang, I.S.; Lee, H.J.; Lee, J.H. General pharmacological profiles of bee venom and its water soluble fractions in rodent models. J. Vet. Sci. 2004, 5, 309–318. [Google Scholar] [PubMed]
- Han, S.; Lee, K.; Yeo, J.; Kweoh, H.; Woo, S.; Lee, M.; Baek, H.; Kim, S.; Park, K. Effect of honey bee venom on microglial cells nitric oxide and tumor necrosis factor-alpha production stimulated by LPS. J. Ethnopharmacol. 2007, 111, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Rekka, E.; Kourounakis, L.; Kourounakis, P. Antioxidant activity of and interleukin production affected by honey bee venom. Arzneimittelforschung 1990, 40, 912–913. [Google Scholar] [PubMed]
- Nam, K.W.; Je, K.H.; Lee, J.H.; Han, H.J.; Lee, H.J.; Kang, S.K.; Mar, W. Inhibition of COX-2 activity and proinflammatory cytokines (TNFalpha and IL-1beta) production by water-soluble sub-fractionated parts from bee (Apis mellifera) venom. Arch. Pharm. Res. 2003, 26, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Kim, S.K.; Han, J.B.; Ahn, H.J.; Bae, H.; Min, B.I. Effects of bee venom on the pro-inflammatory responses in RAW264.7 macrophage cell line. J. Ethnopharmacol. 2005, 99, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Doo, A.R.; Kim, S.N.; Kim, S.T.; Park, J.Y.; Chung, S.H.; Choe, B.Y.; Chae, Y.; Lee, H.; Yin, C.S.; Park, H.J. Bee venom protects SH-SY5Y human neuroblastoma cells from 1-methyl-4-phenylpyridinium-induced apoptotic cell death. Brain Res. 2012, 1429, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Namaka, M.; Crook, A.; Doupe, A.; Kler, K.; Vaconcelos, M.; Klowak, M.; Gong, Y.; Wojewnik-Smith, A.; Melanson, M. Examining the evidence: Complementary adjunctive therapies for multiple sclerosis. Neurol. Res. 2008, 30, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Apel, A.; Greim, B.; Zetti, U.K. How frequently do patients with multiple sclerosis use complementary and alternative medicine? Complement. Ther. Med. 2005, 13, 258–263. [Google Scholar] [CrossRef] [PubMed]
- The American Apitherapy Society Inc. Available online: http://www.apitherapy.org (accessed on 13 April 2015).
- Bowling, A.C. Complementary and alternative medicine and multiple sclerosis. Neurol. Clin. 2011, 29, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Wesselius, T.; Heersema, D.J.; Mostert, J.P.; Heerings, M.; Admiraal-Behloul, F.; Talebian, A.; van Buchem, M.A.; de Keyser, J. A randomized crossover study of bee sting therapy for multiple sclerosis. Neurology 2005, 65, 1764–1768. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A.; Daguio, M.; Wester, D.; Hauser, M.; Kirchman, A.; Skinkis, C. Bee-venom therapy for treating multiple sclerosis - a clinical trial. Altern. Complement. Ther. 2001, 7, 37–45. [Google Scholar] [CrossRef]
- Yang, E.J.; Choi, S.M. Synuclein modification in an ALS animal model. Evid. Based Complement Altern. Med. 2013, 2013, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jung, S.Y.; Lee, K.W.; Lee, S.H.; Cai, M.; Choi, S.M.; Yang, E.J. Bee venom effects on ubiquitin proteasome system in hSOD1(G85R)-expressing NSC34 motor neuron cells. BMC Complement. Altern. Med. 2013, 13, 1–9. [Google Scholar]
- Cunha, A.O.S.; Mortari, M.R.; Oliveira, L.; Carolino, R.O.G.; Coutinho-Netto, J.; Santos, W.F. Anticonvulsant effects of the wasp Polybia ignobilis venom on chemically induced seizures and action on GABA and glutamate receptors. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2005, 141, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Mortari, M.R.; Cunha, A.O.S.; Oliveira, L.; Vieira, E.B.; Gelfuso, E.A.; Coutinho-Netto, J.; Santos, W.F. Anticonvulsant and behavioural effects of the denatured venom of the social wasp Polybia occidentalis (Polistinae, Vespidae). Basic Clin. Pharmacol. Toxicol. 2005, 97, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Couto, L.L.; dos Anjos, L.C.; Araujo, M.A.F.; Mourão, C.A.; Schwartz, C.A.; Ferreira, L.B.; Mortari, M.R. Anticonvulsant and anxiolytic activity of the peptide fraction isolated from the venom of the social wasp Polybia paulista. Pharmacogn. Mag. 2012, 8, 292–299. [Google Scholar] [PubMed]
- Danielisová, V.; Gottlieb, M.; Némethová, M.; Burda, J. Effects of bradykinin postconditioning on endogenous antioxidant enzyme activity after transient forebrain ischemia in rat. Neurochem. Res. 2008, 33, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Danielisová, V.; Gottlieb, M.; Némethová, M.; Kravcuková, P.; Domoráková, I.; Mechírová, E.; Burda, J. Bradykinin postconditioning protects pyramidal CA1 neurons against delayed neuronal death in rat hippocampus. Cell. Mol. Neurobiol. 2009, 29, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, A.B.; Fontana, A.C.K.; Coutinho-Netto, J.; Santos, W.F. Effects of the crude venom of social wasp Agelaia vicina on γ-Aminobutyric acid and glutamate uptake in synapsosomes from rat cerebral cortex. J. Biochem. Mol. Toxicol. 2000, 14, 88–94. [Google Scholar] [PubMed]
- Krzyzanowska, W.; Pomierny, B.; Filip, M.; Pera, J. Glutamate transporters in brain ischemia: To modulate or not? Acta Pharmacol. Sin. 2014, 35, 444–462. [Google Scholar] [CrossRef] [PubMed]
- Soni, N.; Reddy, B.V.; Kumar, P. GLT-1 transporter: An effective pharmacological target for various neurological disorders. Pharmacol. Biochem. Behav. 2014, 127, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Son, M.; Choi, J.; Jun, J.; Kim, J.; Lee, M. Bee venom acupuncture for rheumatoid arthritis: A systematic review of randomised clinical trials. BMJ 2014, 4, e006140. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Giralt, E. Three valuable peptides from bee and wasp venoms for therapeutic and biotechnological use: Mellitin, Apamin and Mastoparan. Toxins 2015, 7, 1126–1150. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Son, D.; Lee, J.; Lee, Y.; Song, H.; Lee, C.; Hong, J. Therapeutic application of anti-arthritis, pain-releasing, anti-cancer effects of bee venom and its constituent compounds. Pharmacol. Ther. 2007, 115, 246–270. [Google Scholar] [CrossRef] [PubMed]
- Vyatchannikov, N.K.; Sinka, A.Y. The effect of mellitin—The major constituent of the bee venom-on the central nervous system. Farmakol. Toksikol. 1973, 36, 526–530. [Google Scholar]
- Ishay, J.; Ben-Shachrar, D.; Elazar, Z.; Kaplinsky, E. Effects of melittin on the central nervous system. Toxicon 1975, 13, 277–283. [Google Scholar] [CrossRef]
- Yang, E.J.; Kim, S.H.; Yang, S.C.; Lee, S.M.; Choi, S.M. Melittin restores proteasome function in an animal model of ALS. J. Neuroinflammation 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- ChemSpider, Melittin. Available online: http://www.chemspider.com/Chemical-Structure.26567345.html (accessed on 11 August 2015).
- ChemSpider, Apamin. Available online: http://www.chemspider.com/Chemical-Structure.21169555.html (accessed on 11 August 2015).
- Toxin and Toxin Target Database, Alpha-pompilidotoxin. Available online: http://www.t3db.ca/toxins/T3D2490 (accessed on 11 August 2015).
- Toxin and Toxin Target Database, Beta-pompilidotoxin. Available online: http://www.t3db.ca/toxins/T3D2491 (accessed on 11 August 2015).
- ChemSpider, Philanthotoxin. Available online: http://www.chemspider.com/Chemical-Structure.103077.html (accessed on 11 August 2015).
- ChemSpider, Bradykinin. Available online: http://www.chemspider.com/Chemical-Structure.388341.html?rid=15413dd8-4d59-4d52-aac6-45e35a46f78d (accessed on 11 August 2015).
- ChemSpider, Transportan. Available online: http://www.chemspider.com/Chemical-Structure.17290614.html?rid=5ed04597-4113-4665-98c5-cef78b91243 (accessed on 11 August 2015).
- Dantas, C.G.; Nunes, T.L.G.M.; Paixão, A.O.; Reis, F.P.; Júnior, W.L.; Cardoso, J.C.; Gramacho, K.P.; Gomes, M.Z. Pharmacological evaluation of bee venom and Melittin. Rev. Bras. Farmacogn. 2014, 24, 67–72. [Google Scholar] [CrossRef]
- Lamy, C.; Goodchild, S.J.; Weatherall, K.L.; Jane, D.E.; Liégeois, J.F.; Seutin, V.; Marrion, N.V. Allosteric block of KCa2 channels by Apamin. J. Biol. Chem. 2010, 285, 27067–27077. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Coleman, N.; Garing, A.L.; Wulff, H. The therapeutic potential of small-conductance KCa2 channels in neurodegenerative and psychiatric diseases. Expert Opin. Ther. Targets 2013, 17, 1203–1220. [Google Scholar] [CrossRef] [PubMed]
- Stackman, R.W.; Hammond, R.S.; Linardatos, E.; Gerlach, A.; Maylie, J.; Adelman, J.P.; Tzounopoulos, T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J. Neurosci. 2002, 22, 10163–10171. [Google Scholar] [PubMed]
- Gati, C.; Mortari, M.; Schwartz, E. Towards Therapeutic Applications of Arthropod Venom K(+)-channel blockers in CNS neurologic diseases involving memory acquisition and storage. J. Toxicol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Adelman, J.P.; Maylie, J.; Sah, P. Small-conductance Ca2+-activated K+ channels: Form and function. Annu. Rev. Physiol. 2012, 74, 245–269. [Google Scholar] [CrossRef] [PubMed]
- Salthun-Lassalle, B.; Hirsch, E.C.; Wolfart, J.; Ruberg, M.; Michel, P.P. Rescue of Mesencephalic Dopaminergic Neurons in Culture by Low-Level Stimulation of Voltage-Gated. J. Neurosci. 2004, 24, 5922–5930. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Bonnet, A.M.; Schüpbach, M. Medicament for Treating Parkinson Disease. U.S. Patent N° US 8357658 B2, 22 January 2013. [Google Scholar]
- Tzounopoulos, T.; Stackman, R. Enhancing Synaptic Plasticity and Memory: A role for small-conductance Ca2+-activated K+ channels. Neuroscientist 2003, 9, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Alkon, D.L.; Etcheberrigaray, R.; Ito, E.; Gibson, G.E. Diagnostic Tests for Alzheimers Disease. U.S. Patent N° 5580748 A, 3 December 1996. Available online: http://www.google.com.ar/patents/US5580748 (accessed on 13 April 2015). [Google Scholar]
- Masters, C.L.; Bush, A.I.; Beyreuther, K.T. Method of Assaying for Alzheimer’s Disease. U.S. Patent N° 5705401 A, 6 January 1998. Available online: http://www.google.com.ar/patents/US5705401 (accessed on 13 April 2015). [Google Scholar]
- Potter, H. Method of Diagnosing and Monitoring a Treatment for Alzheimer’s Disease. Patent N° 5778893 A, 14 July 1998. Available online: http://www.google.com/patents/US5778893 (accessed on 13 April 2015). [Google Scholar]
- Garcia, M.L.; Galvez, A.; Garcia-Calvo, M.; King, V.F.; Vazquez, J.; Kaczorowski, G.J. Use of toxins to study potassium channels. J. Bioenerg. Biomembr. 1991, 23, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Van der Staay, F.J.; Fanelli, R.J.; Blokland, A.; Schmidt, B.H. Behavioral effects of Apamin, a selective inhibitor of the SKCa-channel, in mice and rats. Neurosci. Biobehav. Rev. 1999, 23, 1087–1110. [Google Scholar] [CrossRef]
- Konno, K.; Miwa, A.; Takayama, H.; Hisada, M.; Itagaki, Y.; Naoki, H.; Yasuhada, T.; Kawai, N. α-Pompilidotoxin (α-PMTX), a novel neurotoxin from the venom of a solitary wasp, facilitates transmission in the crustacean neuromuscular synapse. Neurosc. Lett. 1997, 238, 99–102. [Google Scholar] [CrossRef]
- Konno, K.; Hisada, M.; Itagaki, Y.; Naoki, H.; Kawai, N.; Miwa, A.; Yasuhara, T.; Takayama, H. Isolation and structure of Pompilidotoxins, novel peptide neurotoxins in solitary wasp venoms. Biochem. Biophys. Res. Commun. 1998, 250, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, E.; Stevens, M.; Zaharenko, A.J.; Konno, K.; Tytgat, J.; Wanke, E. Voltage-gated sodium channel isoform-specific effects of Pompilidotoxins. FEBS J. 2010, 277, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Harsch, A.; Konno, K.; Takayama, H.; Kawai, N.; Robinson, H. Effects of α-pompilidotoxin on synchronized firing in networks of rat cortical neurons. Neurosc. Lett. 1998, 252, 49–52. [Google Scholar] [CrossRef]
- Sahara, Y.; Gotoh, M.; Konno, K.; Miwa, A.; Tsubokawa, H.; Robinson, H.P.; Kawai, N. A new class of neurotoxin from wasp venom slows inactivation of sodium current. Eur. J. Neurosci. 2000, 12, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Magloire, V.; Czarnecki, A.; Anwander, H.; Streit, J. β-pompilidotoxin modulates spontaneous activity and persistent sodium currents in spinal networks. Neuroscience 2011, 172, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Yokota, H.; Tsubokawa, H.; Miyawaki, T.; Konno, K.; Nakayama, H.; Masuzawa, T.; Kawai, N. Modulation of synaptic transmission in hippocampal CA1 neurons by a novel neurotoxin (β-pompilidotoxin) derived from wasp venom. Neurosci. Res. 2001, 41, 365–371. [Google Scholar] [CrossRef]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef]
- Eijkelkamp, N.; Linley, J.E.; Baker, M.D.; Minett, M.S.; Cregg, R.; Werdehausen, R.; Rugiero, F.; Wood, J.N. Neurological perspectives on voltage-gated sodium channels. Brain 2012, 135, 2585–2612. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, A.B.; Beleboni, R.O.; Fontana, A.C.; Ribeiro, A.M.; Miranda, A.; Coutinho-Netto, J.; dos Santos, W.F. Characterization of the actions of AvTx 7 isolated from Agelaia vicina (Hymenoptera: Vespidae) wasp venom on synaptosomal glutamate uptake and release. J. Biochem. Mol. Toxicol. 2004, 18, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium channels: Structures, diseases and modulators. Chem. Biol. Drug Des. 2014, 83, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.B.; Ravnborg, M.; Dalgas, U.; Stenager, E. 4-Aminopyridine for symptomatic treatment of multiple sclerosis: A systematic review. Ther. Adv. Neurol. Disord. 2014, 7, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.A.; Stefoski, D.; Rush, J. Orally administered 4-aminopyridine improves clinical signs in multiple sclerosis. Ann. Neurol. 1990, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Luca, C.C.; Singer, C. Can 4-aminopyridine modulate dysfunctional gait networks in Parkinson’s Disease? Parkinsonism Relat. Disord. 2013, 19, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Hirai, Y.; Yasuhara, T.; Yoshida, H.; Nakajima, T.; Fujino, M.; Kitada, C. A new mast cell degranulatin peptide “mastoparano” in the venom of Vespula lewisii. Chem. Pharm. Bull. 1979, 27, 1942–1944. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, P.S.; Garzon, J. Mastoparan reduces the supraspinal analgesia mediated by lX/6-opioid receptors in mice. Eur. J. Pharm. 1994, 258, 159–162. [Google Scholar] [CrossRef]
- Blazquez, P.S.; Garzon, J. αN-AcetyI-β-Endorphin-(1_31) Disrupts the Diminishing Effect of Mastoparan on Opioid- and Clonidine-Evoked Supraspinal Antinociception in Mice. JPET 1995, 273, 787–792. [Google Scholar]
- Yandek, L.E.; Pokomy, A.; Floren, A.; Knoelke, K.; Langel, U.; Almeida, P.F.F. Mechanism of the Cell-Penetrating Peptide Transportan 10 permeation of lipid bilayers. Biophys. J. 2007, 92, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Rocha, T.; Souza, B.M.; Palma, M.S.; Cruz-Höfling, M.A.; Harris, J.B. The neurotoxicological effects of mastoparano Polybia-MPII at the murine neuromuscular junction: An ultrastructural and immunocytochemical study. Histochem. Cell Biol. 2009, 132, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ray, R.; Singh, B.R.; Ray, P. Mastoaparan-7 rescues botulinum toxin-A poisoned neurons in a mause spinal cord cell culture model. Toxicon 2013, 76, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.M.; Cabrera, M.P.S.; Neto, J.R.; Palma, M.S. Investigating the effect of different positioning of lysine residues along the peptide chain of mastoparans for their secondary structures and biological activities. Amino Acids 2011. [Google Scholar] [CrossRef]
- Silva, A.V.R.; Souza, B.M.; Cabrera, M.P.S.; Dias, N.B.; Gomes, P.C.; Neto, J.R.; Stabeli, R.G.; Palma, M.S. The effects of C-terminal amidation of mastoparans on their biological actions and interactions with membrane-mimetic systems. Biochim. Biophys. Acta 2014, 1838, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Fanghänel, S.; Wadhwani, P.; Strandberg, E.; Verdurmen, W.P.; Bürck, J.; Ehni, S.; Mykhailiuk, P.K.; Afonin, S.; Gerthsen, D.; Komarov, I.V.; et al. Structure analysis and conformational transitions of the cell penetrating peptide transportan 10 in the membrane-bound state. PLoS ONE 2014, 9, e99653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, L. Modern methods for delivery drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, U. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [PubMed]
- Yandek, L.E.; Pokorny, A.; Florén, A.; Knoelke, K.; Langel, U.; Almeida, P.F. Mechanism of the cell-penetrating peptide transportan 10 permeation of lipid bilayers. Biophys. J. 2007, 92, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Webling, K.E.; Runesson, J.; Bartfai, T.; Langel, U. Galanin receptors and ligands. Front. Endocrinol. (Lausanne) 2012, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; He, B.; Che, S.; Ginsberg, S.D.; Mufson, E.J. Galanin hyperinnervation upregulates choline acetyltransferase expression in cholinergic basal forebrain neurons in Alzheimer’s disease. Neurodegener. Dis. 2008, 5, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Schiavo, G. Mechanism of action of tetanus and botulinum neurotoxins. Mol. Microbiol. 1995, 13, 1–8. [Google Scholar] [CrossRef]
- Simpson, L. The life story of a botulinum toxin molecule. Toxicon 2013, 68, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Howl, J. Biological applications of the receptor mimetic peptide Mastoparan. Curr. Protein Pept. Sci. 2006, 7, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Todokoro, Y.; Yumen, I.; Fukushima, K.; Kamg, S.; Park, J.; Kohno, T.; Wakamatsu, K.; Akutsu, H.; Fujiwara, T. Structure of tightly membrane-bound Mastoparan-X, a G-protein-activating peptide, determined by solid-state NMR. Biophys. J. 2006, 91, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Higashijima, T.; Uzu, S.; Nakajima, T.; Ross, E.M. Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-biding regulatory proteins (G proteins). JBC 1988, 263, 6491–6494. [Google Scholar]
- Lagerström, M.C.; Schiöth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Thathiah, A.; De Strooper, B. The role of G protein-coupled receptors in the pathology of Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Guixa-Gonzalez, R.; Bruno, A.; Marti-Solano, M.; Selent, J. Crosstalk within GPCR heteromers in schizophrenia and Parkinson’s Disease: Physical or just functional? Curr. Med. Chem. 2012, 19, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Rodríguez-Puertas, R.; Meana, J.J.; García-Sevilla, J.A.; Guimón, J. Neurotransmitter receptor-mediated activation of G-proteins in brains of suicide victims with mood disorders: Selective supersensitivity of alpha(2A)-adrenoceptors. Mol. Psychiatry 2002, 7, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Uzu, S.; Wakamatsu, K.; Saito, K.; Miyazawa, T.; Yasuhara, T.; Tsukamoto, Y.; Fujino, M. Amphiphilic peptides in wasp venom. Biopolymers 1986, 25, S115–S121. [Google Scholar] [PubMed]
- Konno, K.; Palma, M.S.; Hitara, I.Y.; Juliano, M.A.; Juliano, L.; Yasuhara, T. Identification of bradykinins in solitary wasp venoms. Toxicon 2002, 40, 309–312. [Google Scholar] [CrossRef]
- Picolo, G.; Hisada, M.; Moura, A.B.; Machado, M.F.; Conceição, I.M.; Melo, R.L.; Oliveira, V.; Lima-Landman, M.T.; Cury, Y.; Konno, K.; et al. Bradykinin-related peptides in the venom of the solitary wasp Cyphononyx fulvognathus. Biochem. Pharmacol. 2010, 79, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Rocha e Silva, M.; Beraldo, W.T.; Rosenfield, G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am. J. Physiol. 1949, 156, 261–273. [Google Scholar] [PubMed]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The kallikrein-kinin system: Current and future pharmacological targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef] [PubMed]
- Piek, T.; Hue, B.; Mony, L.; Nakajima, T.; Pelhate, M.; Yasuhara, T. Block of synaptic transmission in insect CNS by toxins from the venom of the wasp Megascolia flavifrons (Fab.). Comp. Biochem. Physiol. C 1987, 87, 287–295. [Google Scholar] [CrossRef]
- Noda, M.; Kariura, Y.; Pannasch, U.; Nishikawa, K.; Wang, L.; Seike, T.; Ifuku, M.; Kosai, Y.; Wang, B.; Nolte, C.; et al. Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia. J. Neurochem . 2007, 101, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Golias, Ch.; Charalabopoulos, A.; Stagikas, D.; Charalabopoulos, K.; Batistatou, A. The kinin system—Bradykinin: Biological effects and clinical implications. Multiple role of the kinin system—Bradykinin. Hippokratia 2007, 11, 124–128. [Google Scholar] [PubMed]
- Thornton, E.; Ziebell, J.M.; Leonard, A.V.; Vink, R. Kinin receptor antagonists as potential neuroprotective agents in central nervous system injury. Molecules 2010, 15, 6598–6618. [Google Scholar] [CrossRef] [PubMed]
- Yasuyoshi, H.; Kashii, S.; Zhang, S.; Nishida, A.; Yamauchi, T.; Honda, Y.; Asano, Y.; Sato, S.; Akaike, A. Protective effect of bradykinin against glutamate neurotoxicity in cultured rat retinal neurons. Investig. Ophtalmol. Vis. Sci. 2000, 41, 2273–2278. [Google Scholar]
- Mortari, M.R.; Cunha, A.O.S.; Carolino, R.O.G.; Coutinho-Netto, J.C.; Tomaz, N.P.; Coimbra, N.C.; Santos, W.F. Inhibition of acute nociceptive responses in rats after i.c.v. injection of Thr6-bradykinin, isolated from the venom of the social wasp, Polybia occidentalis. BJP 2007, 151, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Mierke, D.F. Threonine6-bradykinin: Molecular dynamics simulations in a biphasic membrane mimetic. J. Med. Chem. 1997, 40, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Strømgaard, K.; Andersen, K.; Krogsgaard-Larsen, P.; Jaroszewski, J.W. Recent Advances in the Medicinal Chemistry of Polyamine Toxins. Mini Rev. Med. Chem. 2001, 1, 317–338. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.F.; Tikhonov, D.B.; Bølcho, U.; Bolshakov, K.; Nelson, J.K.; Pluteanu, F.; Mellor, R.; Egebjerg, J.; Strømgaard, K. Uncompetitive Antagonism of AMPA Receptors: Mechanistic Insights from Studies of Polyamine Toxin Derivatives. J. Med. Chem. 2006, 49, 5414–5423. [Google Scholar] [CrossRef] [PubMed]
- Eldefrawi, A.T.; Eldefrawi, M.E.; Konno, K.; Mansour, N.A.; Nakanishi, K.; Oltz, E.; Usherwood, P.N.R. Structure and synthesis of a potent glutamate receptor antagonist in wasp venom. Proc. Natl. Acad. Sci. USA 1988, 85, 4910–4913. [Google Scholar] [CrossRef] [PubMed]
- Mellor, I.R.; Usherwood, P.N.R. Targeting ionotropic receptors with polyamine-containing toxins. Toxicon 2004, 43, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Strømgaard, K.; Jensen, L.S.; Vogensen, S.B. Polyamine toxins: Development of selective ligands for ionotropic receptors. Toxicon 2005, 45, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Strømgaard, K.; Mellor, I. AMPA Receptor Ligands: Synthetic and Pharmacological Studies of Polyamines and Polyamine Toxins. Med. Res. Rev. 2004, 24, 589–620. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Kasper, B.; Yuan, H.H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, D.; Jiang, A.; Taly, A.; Chataigneau, R.; Specht, A.; Grutter, T. Ligand-Gated Ion Channels: New Insights into Neurological Disorders and Ligand Recognition. Chem. Rev. 2012, 112, 6285–6318. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.H.; Simon, L.; Strømgaard, K.; Kristensen, A.S. Inhibition of AMPA Receptors by Polyamine Toxins is Regulated by Agonist Efficacy and Stargazin. Neurochem. Res. 2014, 39, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Pathologically activated therapeutics for neuroprotection. Nat. Rev. Neurosci. 2007, 8, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Glasgow, N.G.; Povysheva, N.V. Recent insights into the mode of action of memantine and ketamine. Curr. Opin. Pharmacol. 2015, 20, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B. Ion channels of glutamate receptors: Structural modeling. Mol. Membr. Biol. 2007, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Nørager, N.G.; Jensen, C.B.; Rathje, M.; Andersen, J.; Madsen, K.L.; Kristensen, A.S.; Strømgaard, K. Development of potent fluorescent polyamine toxins and application in labeling of ionotropic glutamate receptors in hippocampal neurons. ACS Chem. Biol. 2013, 8, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.; Monge-Fuentes, V.; Gomes, F.; Lopes, K.; Anjos, L.D.; Campos, G.; Arenas, C.; Biolchi, A.; Gonçalves, J.; Galante, P.; et al. Pharmacological Alternatives for the Treatment of Neurodegenerative Disorders: Wasp and Bee Venoms and Their Components as New Neuroactive Tools. Toxins 2015, 7, 3179-3209. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7083179
Silva J, Monge-Fuentes V, Gomes F, Lopes K, Anjos LD, Campos G, Arenas C, Biolchi A, Gonçalves J, Galante P, et al. Pharmacological Alternatives for the Treatment of Neurodegenerative Disorders: Wasp and Bee Venoms and Their Components as New Neuroactive Tools. Toxins. 2015; 7(8):3179-3209. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7083179
Chicago/Turabian StyleSilva, Juliana, Victoria Monge-Fuentes, Flávia Gomes, Kamila Lopes, Lilian Dos Anjos, Gabriel Campos, Claudia Arenas, Andréia Biolchi, Jacqueline Gonçalves, Priscilla Galante, and et al. 2015. "Pharmacological Alternatives for the Treatment of Neurodegenerative Disorders: Wasp and Bee Venoms and Their Components as New Neuroactive Tools" Toxins 7, no. 8: 3179-3209. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7083179