In Silico Structural and Functional Characterization of HtrA Proteins of Leptospira spp.: Possible Implications in Pathogenesis

 ,
,  and
and 
Abstract
:1. Introduction
2. Methods
2.1. HtrAs Sequence Analysis Among Leptospira spp. and Functional Characterization
2.2. Protein Alignment
2.3. Three-Dimensional (3D) Structure and the Search for Conserved Domains
3. Results
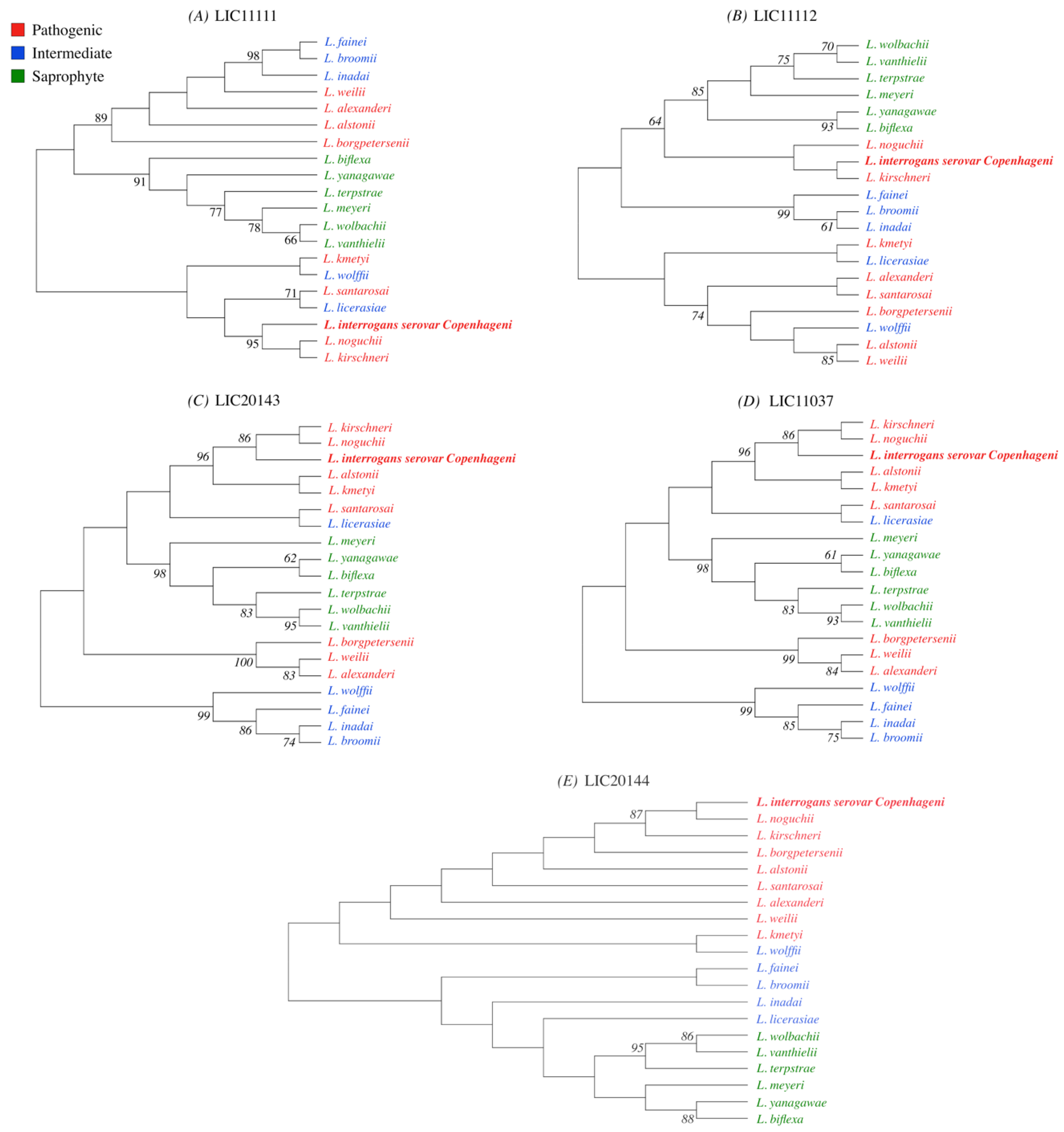
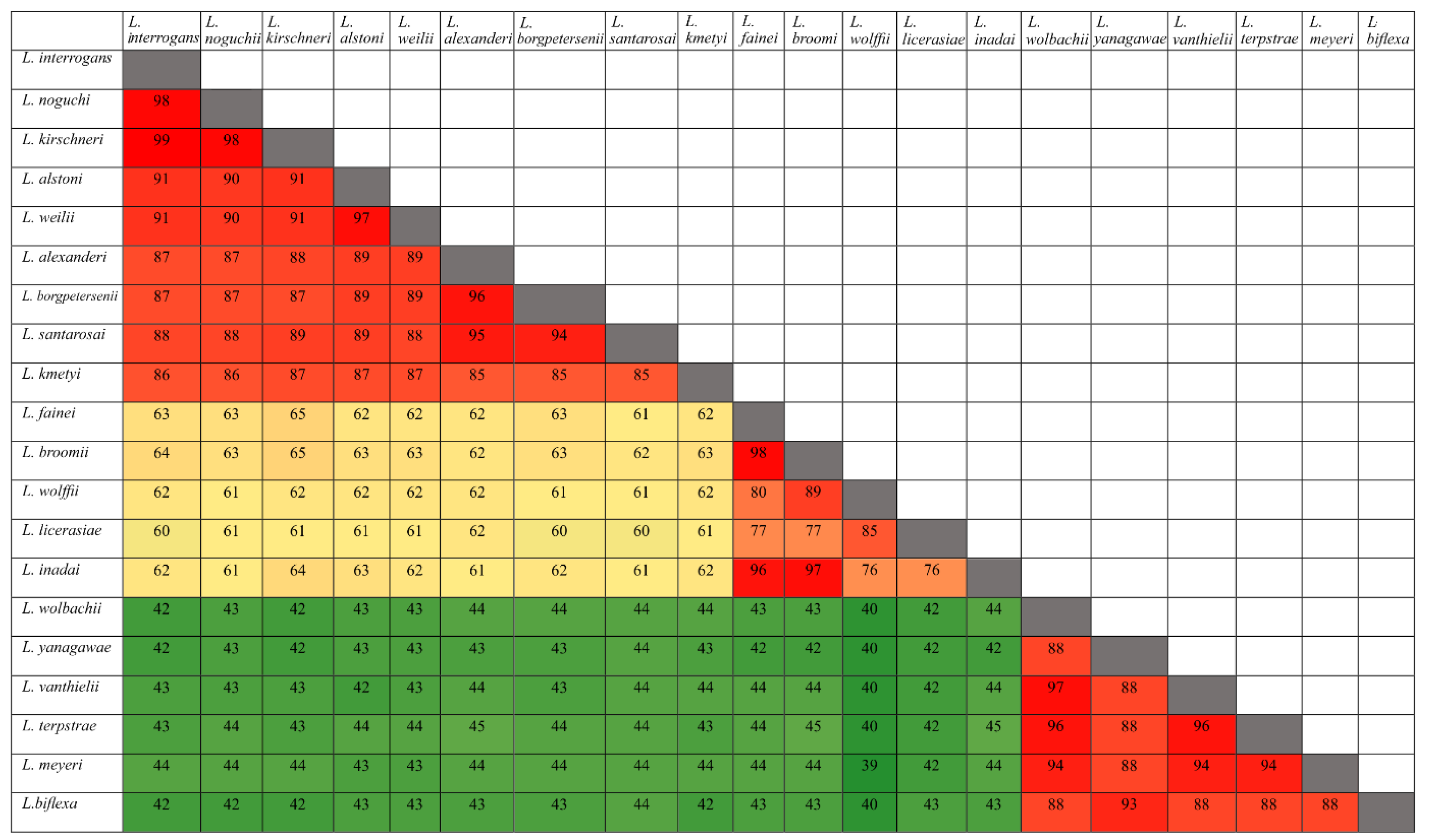
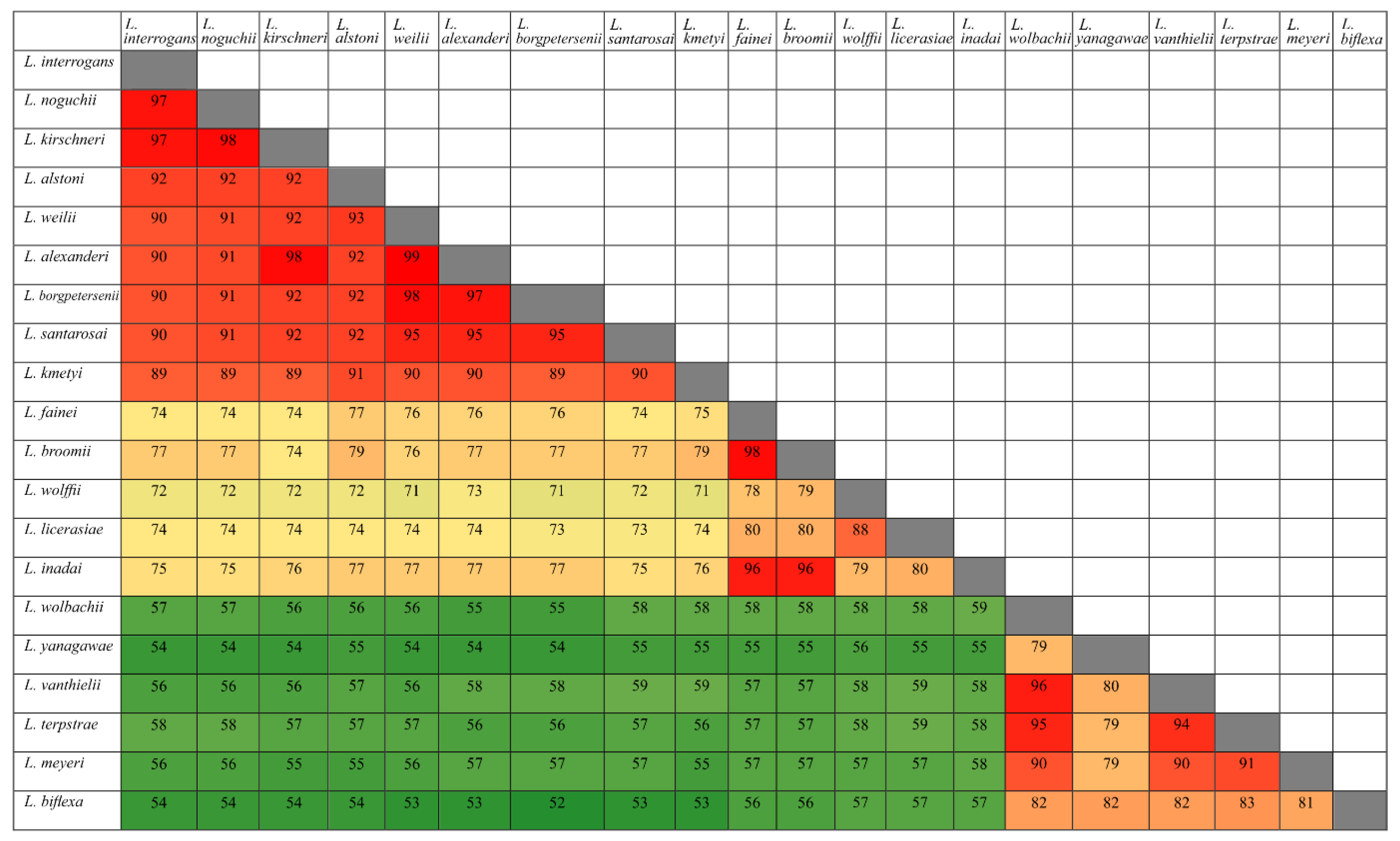
3.1. Conservation Analysis in Leptospiral Strains
3.2. Distribution of HtrAs of Pathogenic, Intermediate, and Saprophytic Leptospira Strains
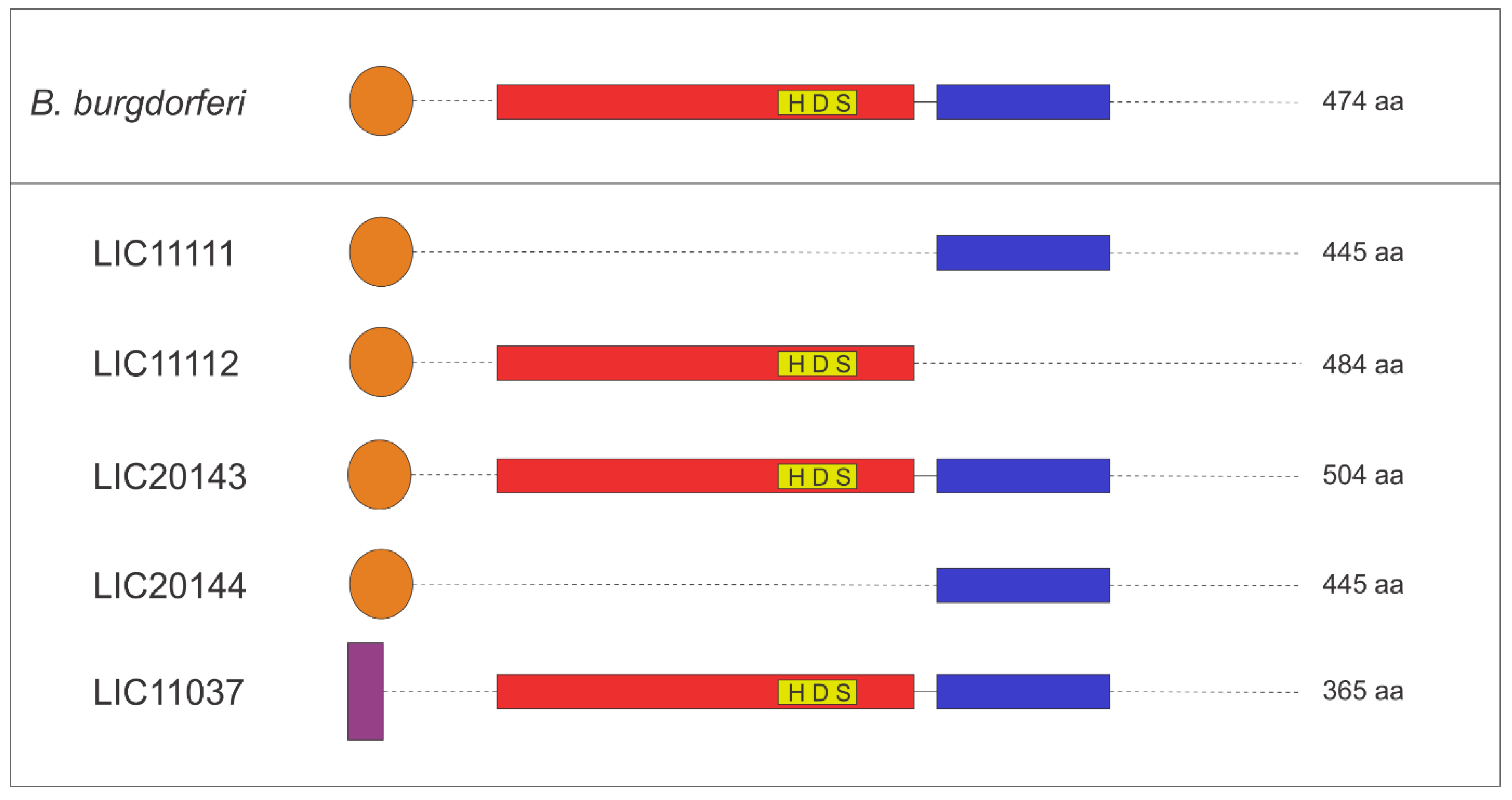
3.3. Features of the HtrA Proteins
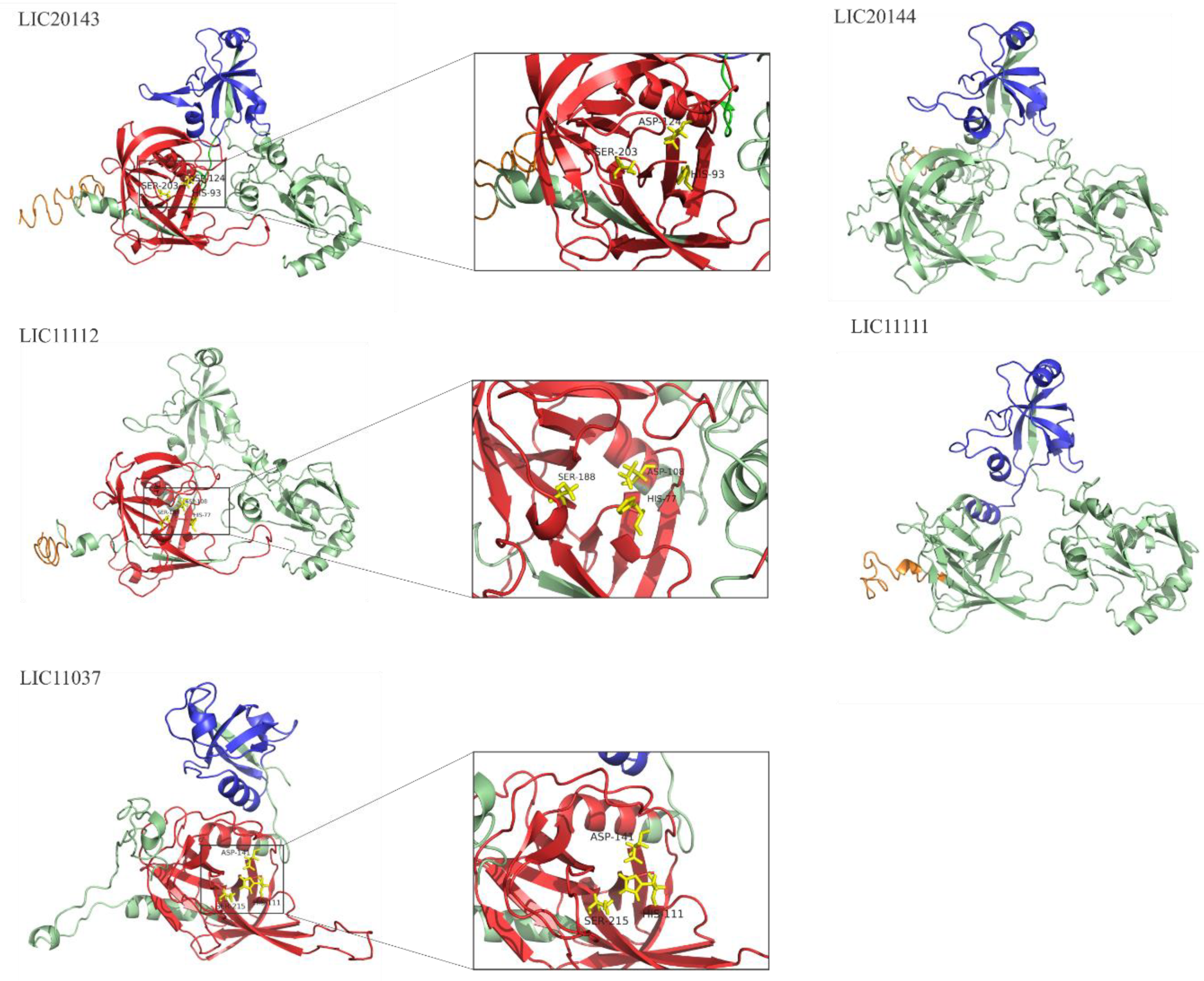
3.4. 3D Structure Model and Domain Architecture
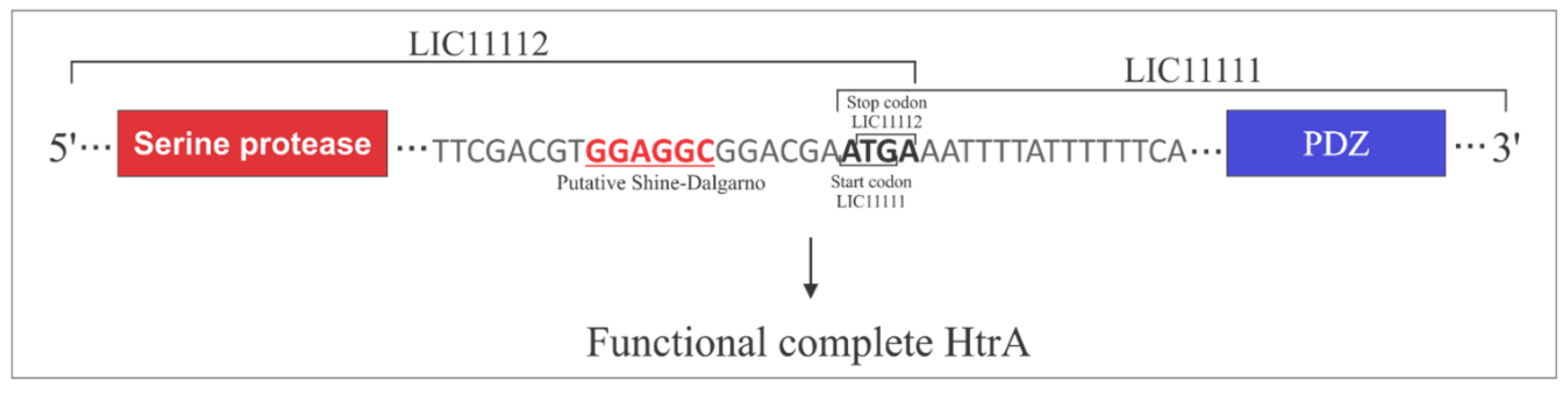
3.5. Operon Properties
3.6. Search for Serine Protease Domain by Alignment
3.7. Serine Protease Domain Alignment for LIC20143, LIC11112, and LIC11037
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Costa, F.; Hagan, J.E.; Calcagno, J.; Kane, M.; Torgerson, P.; Martinez-Silveira, M.S.; Stein, C.; Abela-Ridder, B.; Ko, A.I. Global Morbidity and Mortality of Leptospirosis: A Systematic Review. PLoS Negl. Trop. Dis. 2015, 9, e0003898. [Google Scholar] [CrossRef] [PubMed]
- Levett, P.N. Leptospirosis. Clin. Microbiol. Rev. 2001, 296–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharti, A.R.; Nally, J.E.; Ricaldi, J.N.; Matthias, M.A.; Diaz, M.M.; Lovett, M.A.; Levett, P.N.; Gilman, R.H.; Willig, M.R.; Gotuzzo, E.; et al. Leptospirosis: A zoonotic disease of global importance. Lancet Infect. Dis. 2003, 3, 757–771. [Google Scholar] [CrossRef]
- Wunder, E.A.; Figueira, C.P.; Santos, G.R.; Lourdault, K.; Matthias, M.A.; Vinetz, J.M.; Ramos, E.; Haake, D.A.; Picardeau, M.; dos Reis, M.G.; et al. Real-time PCR reveals rapid dissemination of Leptospira interrogans after intraperitoneal and conjunctival inoculation of hamsters. Infect. Immun. 2016, 84, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Faine, S.; Adler, B.; Bolin, C.; Perolat, P. Leptospira and Leptospirosis, 2nd ed.; MediSci: Melbourne, Australia, 1999. [Google Scholar]
- Picardeau, M. Virulence of the zoonotic agent of leptospirosis: Still terra incognita? Nat. Rev. Microbiol. 2017, 15, 297–307. [Google Scholar] [CrossRef]
- de Faria, M.T.; Calderwood, M.S.; Athanazio, D.A.; McBride, A.J.A.; Hartskeerl, R.A.; Pereira, M.M.; Ko, A.I.; Reis, M.G. Carriage of Leptospira interrogans among domestic rats from an urban setting highly endemic for leptospirosis in Brazil. Acta Trop. 2008, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, A.L.T.O.; Verjovski-Almeida, S.; Van Sluys, M.A.; Monteiro-Vitorello, C.B.; Camargo, L.E.A.; Digiampietri, L.A.; Harstkeerl, R.A.; Ho, P.L.; Marques, M.V.; Oliveira, M.C.; et al. Genome features of Leptospira interrogans serovar Copenhageni. Brazilian J. Med. Biol. Res. 2004, 37, 459–477. [Google Scholar] [CrossRef] [Green Version]
- Vieira, M.L.; Alvarez-Flores, P.; Kirchgatter, K.; Romero, E.C.; Alves, I.J.; De Morais, Z.M.; Vasconcellos, S.A.; Chudzinski-Tavassi, A.M.; Nascimento, A.L.T.O. Interaction of leptospira interrogans with human proteolytic systems enhances dissemination through endothelial cells and protease levels. Infect. Immun. 2013, 81, 1764–1774. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, R.; Domingos, R.F.; Siqueira, G.H.; Fernandes, L.G.; Souza, N.M.; Vieira, M.L.; de Morais, Z.M.; Vasconcellos, S.A.; Nascimento, A.L.T.O. Adhesins of Leptospira interrogans Mediate the Interaction to Fibrinogen and Inhibit Fibrin Clot Formation In Vitro. PLoS Negl. Trop. Dis. 2013, 7, e2396. [Google Scholar] [CrossRef] [Green Version]
- Kassegne, K.; Hu, W.; Ojcius, D.M.; Sun, D.; Ge, Y.; Zhao, J.; Yang, X.F.; Li, L.; Yan, J. Identification of collagenase as a critical virulence factor for invasiveness and transmission of pathogenic leptospira species. J. Infect. Dis. 2014, 209, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Janwitthayanan, W.; Keelawat, S.; Payungporn, S.; Lowanitchapat, A.; Suwancharoen, D.; Poovorawan, Y.; Chirathaworn, C. In vivo gene expression and immunoreactivity of Leptospira collagenase. Microbiol. Res. 2013, 168, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Chura-Chambi, R.M.; Fraga, T.R.; da Silva, L.B.; Yamamoto, B.B.; Isaac, L.; Barbosa, A.S.; Morganti, L. Leptospira interrogans thermolysin refolded at high pressure and alkaline pH displays proteolytic activity against complement C3. Biotechnol. Rep. 2018, 19, e00266. [Google Scholar] [CrossRef] [PubMed]
- Amamura, T.A.; Fraga, T.R.; Vasconcellos, S.A.; Barbosa, A.S.; Isaac, L. Pathogenic Leptospira secreted proteases target the membrane attack complex: A potential role for thermolysin in complement inhibition. Front. Microbiol. 2017, 8, 958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, T.R.; Courrol, D.D.S.; Castiblanco-Valencia, M.M.; Hirata, I.Y.; Vasconcellos, S.A.; Juliano, L.; Barbosa, A.S.; Isaac, L. Immune evasion by pathogenic Leptospira strains: The secretion of proteases that directly cleave complement proteins. J. Infect. Dis. 2014, 209, 876–886. [Google Scholar] [CrossRef] [Green Version]
- da Silva, L.B.; Menezes, M.C.; Kitano, E.S.; Oliveira, A.K.; Abreu, A.G.; Souza, G.O.; Heinemann, M.B.; Isaac, L.; Fraga, T.R.; Serrano, S.M.T.; et al. Leptospira interrogans secreted proteases degrade extracellular matrix and plasma proteins from the host. Front. Cell. Infect. Microbiol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Anu, P.V.; Madanan, M.G.; Nair, A.J.; Nair, G.A.; Nair, G.P.M.; Sudhakaran, P.R.; Satheeshkumar, P.K. Heterologous Expression, Purification and Characterization of an Oligopeptidase A from the Pathogen Leptospira interrogans. Mol. Biotechnol. 2018, 60, 302–309. [Google Scholar] [CrossRef]
- Ingmer, H.; Brøndsted, L. Proteases in bacterial pathogenesis. Res. Microbiol. 2009, 160, 704–710. [Google Scholar] [CrossRef]
- Gottesman, S. Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 1996, 30, 465–506. [Google Scholar] [CrossRef]
- Pallen, M.J.; Wren, B.W. The HtrA family of serine proteases. Mol. Microbiol. 1997, 26, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. Htra proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011, 12, 152–162. [Google Scholar] [CrossRef]
- Frees, D.; Brøndsted, L.; Ingmer, H. Bacterial proteases and virulence. Subcell. Biochem. 2013, 66, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Harrer, A.; Boehm, M.; Backert, S.; Tegtmeyer, N. Overexpression of serine protease HtrA enhances disruption of adherens junctions, paracellular transmigration and type IV secretion of CagA by Helicobacter pylori. Gut Pathog. 2017, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Hoy, B.; Rohde, M.; Tegtmeyer, N.; Bæk, K.T.; Oyarzabal, O.A.; Brøndsted, L.; Wessler, S.; Backert, S. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: Role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 2012, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löwer, M.; Weydig, C.; Metzler, D.; Reuter, A.; Starinzki-Powitz, A.; Wessler, S.; Schneider, G. Prediction of extracellular proteases of the human pathogen Helicobacter pylori reveals proteolytic activity of the Hp1018/19 protein HtrA. PLoS ONE 2008, 3, e3510. [Google Scholar] [CrossRef]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [Green Version]
- Russell, T.M.; Delorey, M.J.; Johnson, B.J.B. Borrelia burgdorferiBbHtrA degrades host ECM proteins and stimulates release of inflammatory cytokines in vitro. Mol. Microbiol. 2013, 90, 241–251. [Google Scholar] [CrossRef]
- Coleman, J.L.; Toledo, A.; Benach, J.L. Borrelia burgdorferi HtrA: Evidence for twofold proteolysis of outer membrane protein p66. Mol. Microbiol. 2016, 99, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.L.; Toledo, A.; Benach, J.L. HtrA of Borrelia burgdorferi leads to decreased swarm motility and decreased production of pyruvate. MBio 2018, 9, e01136-18. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, A.L.T.O.; Ko, A.I.; Martins, E.A.L.; Monteiro-Vitorello, C.B.; Ho, P.L.; Haake, D.A.; Verjovski-Almeida, S.; Hartskeerl, R.A.; Marques, M.V.; Oliveira, M.C.; et al. Comparative Genomics of Two Leptospira interrogans Serovars Reveals Novel Insights into Physiology and Pathogenesis. J. Bacteriol. 2004, 186, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-S.; Lin, C.-J.; Hwang, J.-K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Ma, Q.; Zhou, C.; Chen, X.; Zhang, H.; Yang, J.; Mao, F.; Lai, W.; Xu, Y. DOOR 2.0: Presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2014, 42, D654–D659. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Pei, J.; Grishin, N.V. PROMALS3D: Multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol. Biol. 2014, 1079, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Fouts, D.E.; Matthias, M.A.; Adhikarla, H.; Adler, B.; Amorim-Santos, L.; Berg, D.E.; Bulach, D.; Buschiazzo, A.; Chang, Y.F.; Galloway, R.L.; et al. What Makes a Bacterial Species Pathogenic?:Comparative Genomic Analysis of the Genus Leptospira. PLoS Negl. Trop. Dis. 2016, 10, e0004403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, M.L. Translational termination-reinitiation in RNA viruses. Biochem. Soc. Trans. 2010, 38, 1558–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamyshev, A.L.; Karamysheva, Z.N.; Yamami, T.; Ito, K.; Nakamura, Y. Transient idling of posttermination ribosomes ready to reinitiate protein synthesis. Biochimie 2004, 86, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lei, L.; Gong, S.; Chen, D.; Flores, R.; Zhong, G. The chlamydial periplasmic stress response serine protease cHtrA is secreted into host cell cytosol. BMC Microbiol. 2011, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Bumann, D.; Aksu, S.; Wendland, M.; Janek, K.; Zimny-Arndt, U.; Sabarth, N.; Meyer, T.F.; Jungblut, P.R. Proteome analysis of secreted proteins of the gastric pathogen Helicobacter pylori. Infect. Immun. 2002, 70, 3396–3403. [Google Scholar] [CrossRef] [Green Version]
- Gherardini, F.C. Borrelia burgdorferi HtrA may promote dissemination and irritation. Mol. Microbiol. 2013, 90, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Backert, S.; Bernegger, S.; Skórko-Glonek, J.; Wessler, S. Extracellular HtrA serine proteases: An emerging new strategy in bacterial pathogenesis. Cell. Microbiol. 2018, 20, e12845. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, L.L.; Radulic, M.; Doric, M.; Abu Kwaik, Y. HtrA homologue of Legionella pneumophila: An indispensable element for intracellular infection of mammalian but not protozoan cells. Infect. Immun. 2001, 69, 2569–2579. [Google Scholar] [CrossRef] [Green Version]
- Cortés, G.; De Astorza, B.; Benedí, V.J.; Albertí, S. Role of the htrA gene in Klebsiella pneumoniae virulence. Infect. Immun. 2002, 70, 4772–4776. [Google Scholar] [CrossRef] [Green Version]
- Flannagan, R.S.; Aubert, D.; Kooi, C.; Sokol, P.A.; Valvano, M.A. Burkholderia cenocepacia requires a periplasmic HtrA protease for growth under thermal and osmotic stress and for survival in vivo. Infect. Immun. 2007, 75, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Li, S.R.; Dorrell, N.; Everest, P.H.; Dougan, G.; Wren, B.W. Construction and characterization of a Yersinia enterocolitica O:8 high-temperature requirement (htrA) isogenic mutant. Infect. Immun. 1996, 64, 2088–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, S.; Stevenson, A.; Bacon, A.; Weinhardt, A.B.; Roberts, M. The alternative sigma factor, σ(E), is critically important for the virulence of Salmonella typhimurium. Infect. Immun. 1999, 67, 1560–1568. [Google Scholar] [CrossRef]
- Purdy, G.E.; Hong, M.; Payne, S.M. Shigella flexneri DegP facilitates IcsA surface expression and is required for efficient intercellular spread. Infect. Immun. 2002, 70, 6355–6364. [Google Scholar] [CrossRef] [Green Version]
- Gloeckl, S.; Ong, V.A.; Patel, P.; Tyndall, J.D.A.; Timms, P.; Beagley, K.W.; Allan, J.A.; Armitage, C.W.; Turnbull, L.; Whitchurch, C.B.; et al. Identification of a serine protease inhibitor which causes inclusion vacuole reduction and is lethal to Chlamydia trachomatis. Mol. Microbiol. 2013, 89, 676–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.L.; Brown, L.L.; Kirkwood-Watts, D.; Warren, T.K.; Lund, S.A.; King, D.S.; Jones, K.F.; Hruby, D.E. Listeria monocytogenes 10403S HtrA is necessary for resistance to cellular stress and virulence. Infect. Immun. 2006, 74, 765–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatfield, S.N.; Strahan, K.; Pickard, D.; Charles, I.G.; Hormaeche, C.E.; Dougan, G. Evaluation of Salmonella typhimurium strains harbouring defined mutations in htrA and aroA in the murine salmonellosis model. Microb. Pathog. 1992, 12, 145–151. [Google Scholar] [CrossRef]
- Ibrahim, Y.M.; Kerr, A.R.; McCluskey, J.; Mitchell, T.J. Control of virulence by the two-component system CiaR/H is mediated via HtrA, a major virulence factor of Streptococcus pneumoniae. J. Bacteriol. 2004, 186, 5258–5266. [Google Scholar] [CrossRef] [Green Version]
- Lipinska, B.; Zylicz, M.; Georgopoulos, C. The HtrA (DegP) protein, essential for Escherichia coli survival at high temperatures, is an endopeptidase. J. Bacteriol. 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, P.R.H.; Sauer, R.T. Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease. J. Bacteriol. 1996, 178, 1146–1153. [Google Scholar] [CrossRef] [Green Version]
- Picardeau, M.; Bulach, D.M.; Bouchier, C.; Zuerner, R.L.; Zidane, N.; Wilson, P.J.; Creno, S.; Kuczek, E.S.; Bommezzadri, S.; Davis, J.C.; et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLoS ONE 2008, 3, e1607. [Google Scholar] [CrossRef] [Green Version]
- Bulach, D.M.; Zuerner, R.L.; Wilson, P.; Seemann, T.; McGrath, A.; Cullen, P.A.; Davis, J.; Johnson, M.; Kuczek, E.; Alt, D.P.; et al. Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc. Natl. Acad. Sci. USA 2006, 39, 14560–14565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.; Faure, G.; Laass, S.; Kolbe, E.; Seitz, K.; Wehrheim, C.; Wolf, Y.I.; Koonin, E.V.; Soppa, J. Translational coupling via termination-reinitiation in archaea and bacteria. Nat. Commun. 2019, 10, 4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, M.L.; Brown, T.D.K.; Brierley, I. Translational termination-re-initiation in viral systems. Biochem. Soc. Trans. 2008, 36, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nourry, C.; Grant, S.G.N.; Borg, J.P. PDZ domain proteins: Plug and play! Sci. STKE 2003, 2003, RE7. [Google Scholar] [CrossRef]
- Hung, A.Y.; Sheng, M. PDZ domains: Structural modules for protein complex assembly. J. Biol. Chem. 2002, 277, 5699–5702. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.S.; Zhang, M. Signaling complex organization by PDZ domain proteins. Neurosignals 2002, 11, 315–321. [Google Scholar] [CrossRef]
- Sheng, M.; Sala, C. PDZ Domains and the Organization of Supramolecular Complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef]
- Van Ham, M.; Hendriks, W. PDZ domains—Glue and guide. Mol. Biol. Rep. 2003, 30, 69–82. [Google Scholar] [CrossRef]
- Zarzecka, U.; Harrer, A.; Zawilak-Pawlik, A.; Skorko-Glonek, J.; Backert, S. Chaperone activity of serine protease HtrA of Helicobacter pylori as a crucial survival factor under stress conditions. Cell Commun. Signal. 2019, 17, 161. [Google Scholar] [CrossRef] [Green Version]
- Kariu, T.; Yang, X.; Marks, C.B.; Zhang, X.; Pal, U. Proteolysis of BB0323 results in two polypeptides that impact physiologic and infectious phenotypes in Borrelia burgdorferi. Mol. Microbiol. 2013, 88, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Spiess, C.; Beil, A.; Ehrmann, M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 1999, 97, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Wonderling, L.D.; Wilkinson, B.J.; Bayles, D.O. The htrA (degP) Gene of Listeria monocytogenes 10403S Is Essential for Optimal Growth under Stress Conditions. Appl. Environ. Microbiol. 2004, 70, 1935–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caimano, M.J.; Sivasankaran, S.K.; Allard, A.; Hurley, D.; Hokamp, K.; Grassmann, A.A.; Hinton, J.C.D.; Nally, J.E. A Model System for Studying the Transcriptomic and Physiological Changes Associated with Mammalian Host-Adaptation by Leptospira interrogans Serovar Copenhageni. PLoS Pathog. 2014, 10, e1004004. [Google Scholar] [CrossRef] [PubMed]
- Malmström, J.; Beck, M.; Schmidt, A.; Lange, V.; Deutsch, E.W.; Aebersold, R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009, 460, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T.; Asche, C.I.; Boehm, M.; Loessner, H.; Figueiredo, C.; et al. Helicobacter pylori Employs a Unique Basolateral Type IV Secretion Mechanism for CagA Delivery. Cell Host Microbe 2017, 22, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Boehm, M.; Haenel, I.; Hoy, B.; Brøndsted, L.; Smith, T.G.; Hoover, T.; Wessler, S.; Tegtmeyer, N. Extracellular secretion of protease HtrA from Campylobacter jejuni is highly efficient and independent of its protease activity and flagellum. Eur. J. Microbiol. Immunol. 2013, 3, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Eshghi, A.; Pappalardo, E.; Hester, S.; Thomas, B.; Pretre, G.; Picardeau, M. Pathogenic Leptospira interrogans exoproteins are primarily involved in heterotrophic processes. Infect. Immun. 2015, 83, 3061–3073. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Locus | Strain | Cleavage Sites | Sequence | Transmembrane Region | Serine Protease | PDZ Domains | Size (aa) | Cellular Localization |
|---|---|---|---|---|---|---|---|---|
| LIC11111 | L. interrogans | 24–25 | VPLKA/ETILV | - | - | 175–280 | 445 | Outer membrane |
| L. borgpetersenii | 31–32 | GPLKA/ETILV | - | - | 182–287 | 452 | Outer membrane | |
| L. licerasiae | 23–24 | ENVEA/KADSE | - | - | 206–293 | 458 | Outer membrane | |
| L. biflexa | 22–23 | SNLIA/EEFED | - | - | - | 454 | Outer membrane | |
| LIC11112 | L. interrogans | 20–21 | FQLSA/QNGNT | - | 55–230 | - | 484 | Outer membrane |
| L. borgpetersenii | 23–24 | FHLPA/QNGNS | Inside (1–4) | 32–230 | - | 482 | Outer membrane | |
| TMhelix (5–24) | ||||||||
| Outside (25–482) | ||||||||
| L. licerasiae | 21–22 | FPVFS/QTNGN | - | - | 489 | Outer membrane | ||
| L. biflexa | 20–21 | HSAFS/QTESE | - | - | 237–338 | 485 | Outer membrane | |
| LIC11037 | L. interrogans | - | Inside (1–6) TMhelix (7–24) Outside (25–365) | 68–256 | 270-353 | 365 | Outer membrane | |
| L. borgpetersenii | - | Inside (1–6) TMhelix (7–26) Outside (27–368) | 71–259 | 273-356 | 368 | Outer membrane | ||
| L. licerasiae | - | Inside (1–8) TMhelix (9–31) Outside (32–366) | 69–257 | 271-354 | 366 | Outer membrane | ||
| L. biflexa | 33–34 | EIRSA/VTKLF | Inside (1–6) TMhelix (7–29) Outside (30–375) | 72-260 | 289–357 | 375 | Inner membrane | |
| LIC20143 | L. interrogans | 38–39 | SGIYA/VENPL | Inside (1–19) TMhelix (20–39) Outside (40–504) | 56–243 | 252–345 | 504 | Outer membrane |
| L. borgpetersenii | - | 54–241 | 258-343 | 502 | Outer membrane | |||
| L. licerasiae | 21–22 | LPLFS/EERSD | 29-225 | 232–327 | 486 | Outer membrane | ||
| L. biflexa | 51–52 | STSHA/EPNGQ | Inside (1–26) TMhelix (27–49) Outside (50–515) | 69-249 | 265–356 | 515 | Outer membrane | |
| LIC20144 | L. interrogans | 19–20 | FEVLA/EISSK | - | - | 263–343 | 527 | Outer membrane |
| L. borgpetersenii | 19–20 | LGVFA/EKVES | - | - | 263–343 | 525 | Outer membrane | |
| L. liceraseae | 21–22 | SGVSA/KKPKP | - | - | 243–323 | 508 | Outer membrane | |
| L. biflexa | - | - | - | - | - | 490 | Cytoplasmatic | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daroz, B.B.; Fernandes, L.G.V.; Teixeira, A.F.; Nascimento, A.L.T.O. In Silico Structural and Functional Characterization of HtrA Proteins of Leptospira spp.: Possible Implications in Pathogenesis. Trop. Med. Infect. Dis. 2020, 5, 179. https://0-doi-org.brum.beds.ac.uk/10.3390/tropicalmed5040179
Daroz BB, Fernandes LGV, Teixeira AF, Nascimento ALTO. In Silico Structural and Functional Characterization of HtrA Proteins of Leptospira spp.: Possible Implications in Pathogenesis. Tropical Medicine and Infectious Disease. 2020; 5(4):179. https://0-doi-org.brum.beds.ac.uk/10.3390/tropicalmed5040179
Chicago/Turabian StyleDaroz, Brenda Bevilaqua, Luis Guilherme Virgílio Fernandes, Aline Florencio Teixeira, and Ana Lucia Tabet Oller Nascimento. 2020. "In Silico Structural and Functional Characterization of HtrA Proteins of Leptospira spp.: Possible Implications in Pathogenesis" Tropical Medicine and Infectious Disease 5, no. 4: 179. https://0-doi-org.brum.beds.ac.uk/10.3390/tropicalmed5040179