Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Clinical Specimens
2.2. Collection of Wastewater Samples
2.3. Viral Concentration and RNA Extraction
2.4. Reverse Transcription PCR (RT-PCR) Amplification and Sequencing
2.5. Norovirus Phylogenetic Analysis
2.6. NGS Data Analysis
2.7. Analysis of Amino Acid Variation within GII.4 Capsid Sequences
2.8. Molecular Adaptation Analysis of the Norovirus GII.4 Capsid
3. Results
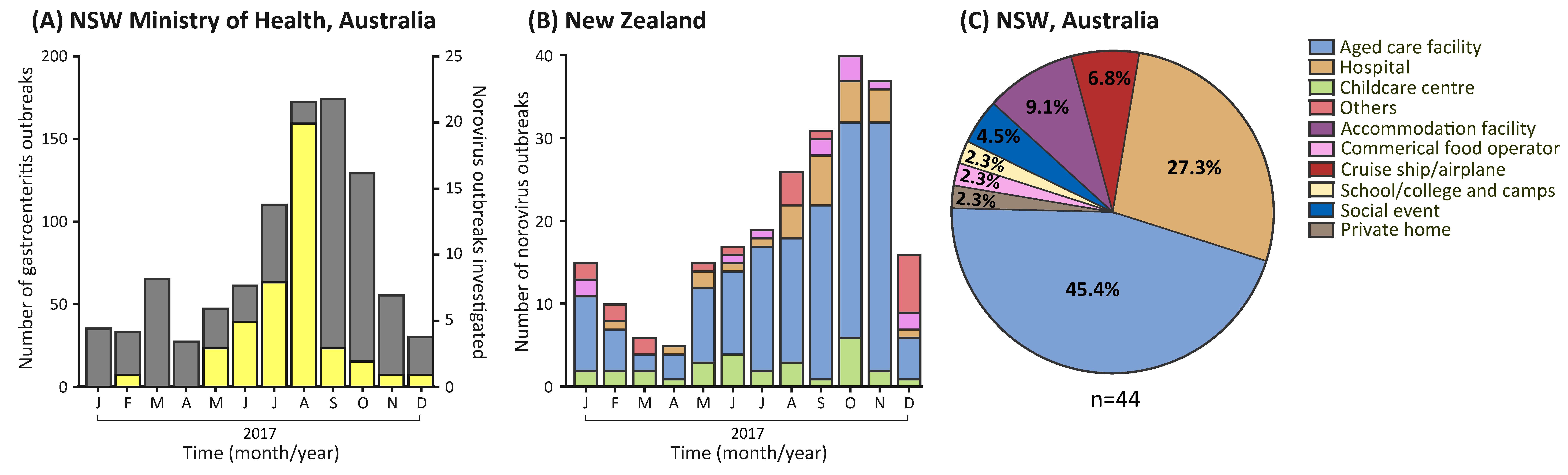
3.1. Gastroenteritis Outbreak Increase in 2017
3.2. Outbreak Settings
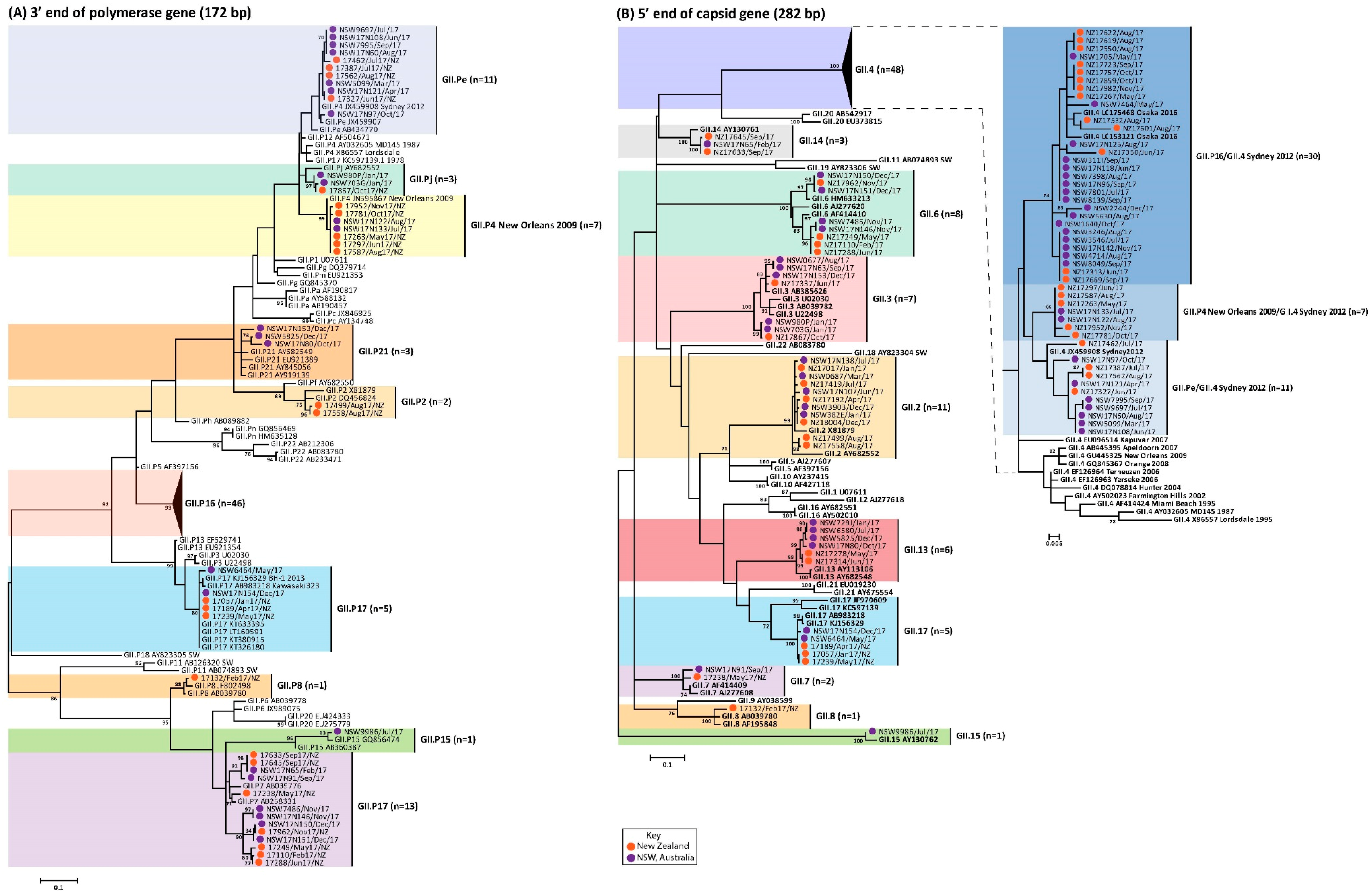
3.3. Circulating GI Noroviruses
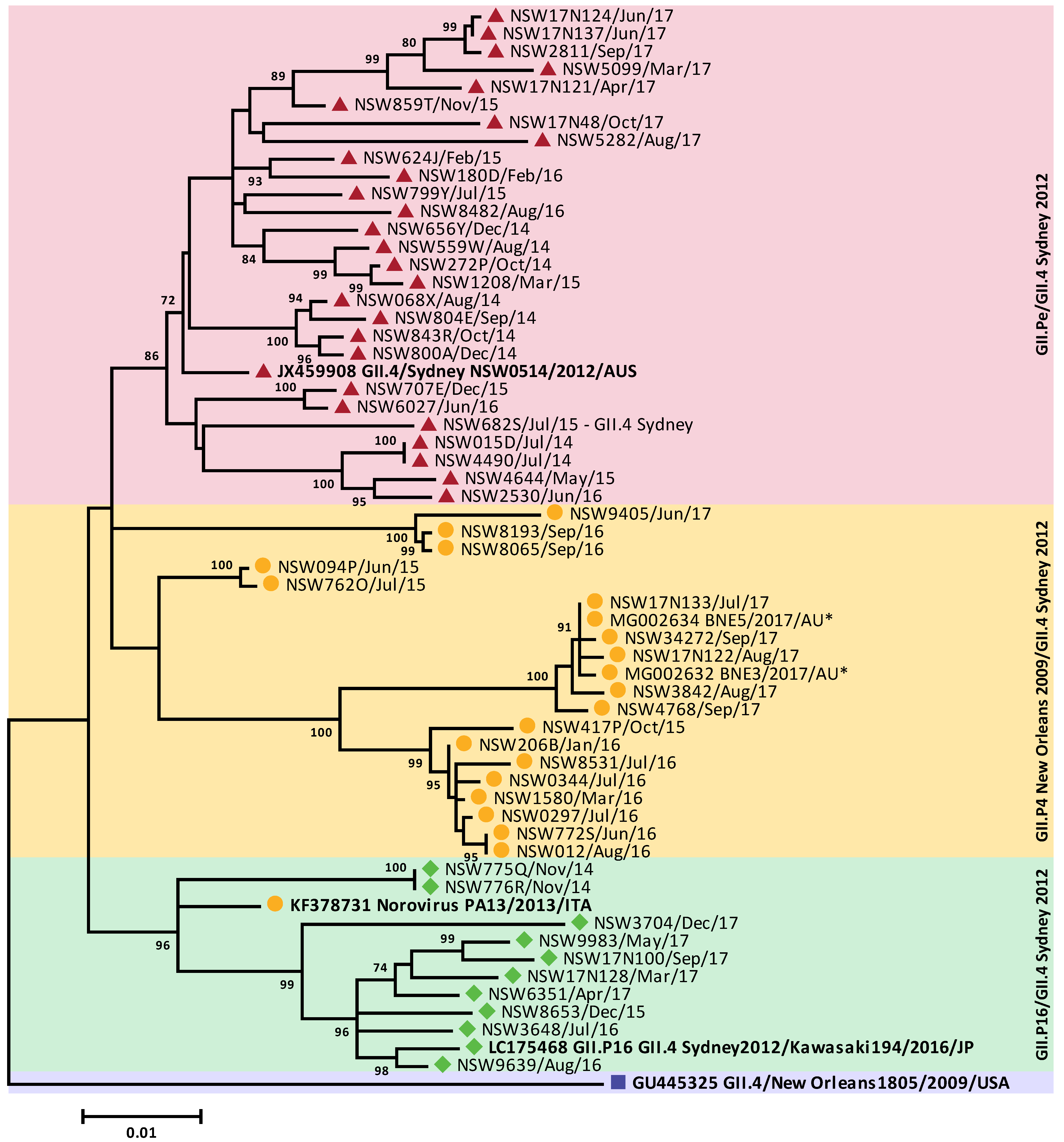
3.4. Circulating GII Noroviruses
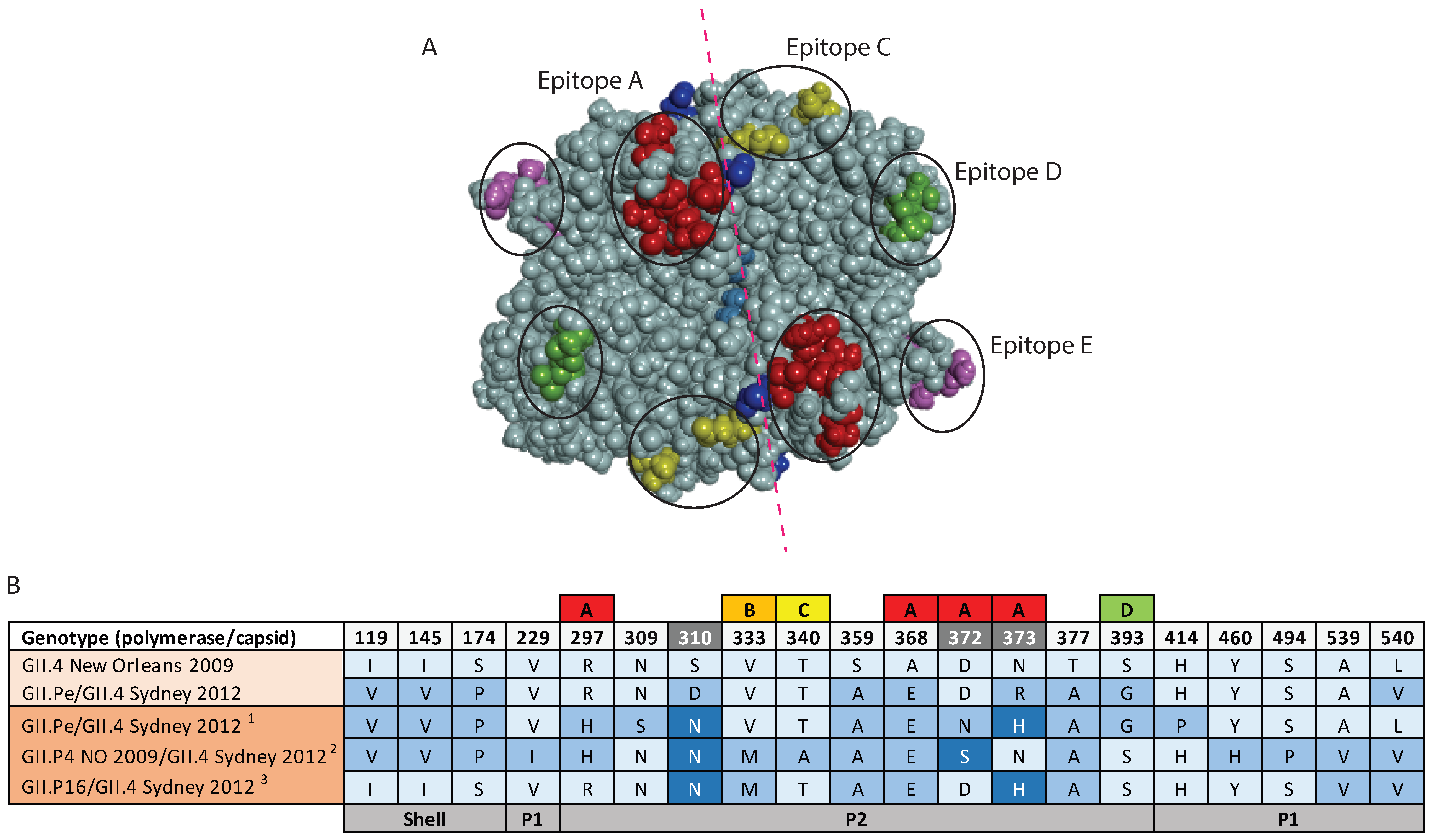
3.5. Antigenic Variation within the GII.4 Capsids
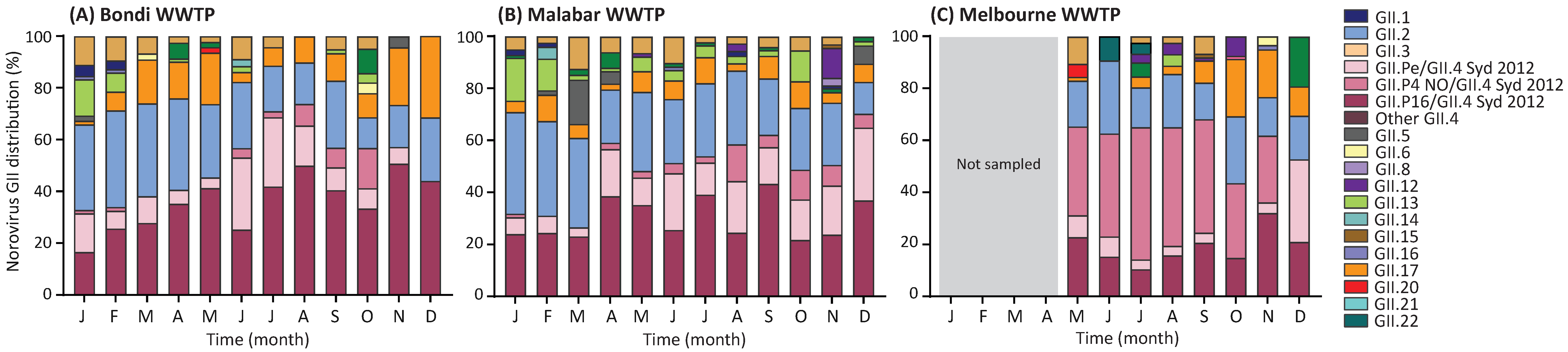
3.6. Norovirus GII Genotype Distribution in Wastewater Samples
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global economic burden of norovirus gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [PubMed]
- Siebenga, J.J.; Vennema, H.; Zheng, D.-P.; Vinjé, J.; Lee, B.E.; Pang, X.-L.; Ho, E.C.; Lim, W.; Choudekar, A.; Broor, S. Norovirus illness is a global problem: Emergence and spread of norovirus GII. 4 variants, 2001–2007. J. Infect. Dis. 2009, 200, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Teunis, P.F.; Moe, C.L.; Liu, P.; Miller, S.E.; Lindesmith, L.; Baric, R.S.; Le Pendu, J.; Calderon, R.L. Norwalk virus: How infectious is it? J. Med. Virol. 2008, 80, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinjé, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, V.; Lorusso, E.; Decaro, N.; Elia, G.; Radogna, A.; D’Abramo, M.; Desario, C.; Cavalli, A.; Corrente, M.; Camero, M. Detection and molecular characterization of a canine norovirus. Emerg. Infect. Dis. 2008, 14, 1306. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, J.R.; Barclay, L.; Nascimento, M.S.J.; Vinjé, J. Novel norovirus in dogs with diarrhea. Emerg. Infect. Dis. 2010, 16, 980. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Donaldson, E.F.; LoBue, A.D.; Cannon, J.L.; Zheng, D.-P.; Vinje, J.; Baric, R.S. Mechanisms of GII. 4 norovirus persistence in human populations. PLoS Med. 2008, 5, e31. [Google Scholar] [CrossRef] [PubMed]
- Shanker, S.; Choi, J.-M.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII. 4 epidemic variant: Implications for epochal evolution. J. Virol. 2011, 85, 8635–8645. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tu, E.T.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Emergence of a new norovirus genotype II. 4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 2006, 44, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.-S.; Hewitt, J.; Lim, K.L.; Boni, M.F.; Merif, J.; Greening, G.; Ratcliff, R.M.; Holmes, E.C.; Tanaka, M.M.; Rawlinson, W.D. The emergence and evolution of the novel epidemic norovirus GII. 4 variant Sydney 2012. Virology 2014, 450, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.; Fankhauser, R.; Ando, T.; Monroe, S.; Glass, R. Identification of a distinct common strain of “Norwalk-like viruses” having a global distribution. J. Infect. Dis. 1999, 179, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Tu, E.T.-V.; Bull, R.A.; Greening, G.E.; Hewitt, J.; Lyon, M.J.; Marshall, J.A.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Epidemics of gastroenteritis during 2006 were associated with the spread of norovirus GII. 4 variants 2006a and 2006b. Clin. Infect. Dis. 2008, 46, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tanaka, M.M.; White, P.A. Norovirus recombination. J. Gen. Virol. 2007, 88, 3347–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, J.-S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the pandemic norovirus GII. 4 lineage. J. Virol. 2013, 87, 6270–6282. [Google Scholar] [CrossRef] [PubMed]
- Lun, J.H.; Hewitt, J.; Sitabkhan, A.; Eden, J.-S.; Tuipulotu, D.E.; Netzler, N.E.; Morrell, L.; Merif, J.; Jones, R.; Huang, B. Emerging recombinant noroviruses identified by clinical and waste water screening. Emerg. Microbes Infect. 2018, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, V.; Medici, M.; De Grazia, S.; Tummolo, F.; Calderaro, A.; Bonura, F.; Saporito, L.; Terio, V.; Catella, C.; Lanave, G. Evidence for recombination between pandemic GII. 4 norovirus strains New Orleans 2009 and Sydney 2012. J. Clin. Microbiol. 2013, 51, 3855–3857. [Google Scholar] [CrossRef] [PubMed]
- Bruggink, L.; Catton, M.; Marshall, J. A norovirus intervariant GII. 4 recombinant in Victoria, Australia, June 2016: The next epidemic variant? Euro Surveill. 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Fonager, J.; Barzinci, S.; Fischer, T. Emergence of a new recombinant Sydney 2012 norovirus variant in Denmark, 26 December 2012 to 22 March 2013. Euro Surveill. 2013, 18, 18. [Google Scholar] [CrossRef]
- Wong, T.N.; Dearlove, B.L.; Hedge, J.; Giess, A.P.; Piazza, P.; Trebes, A.; Paul, J.; Smit, E.; Smith, E.G.; Sutton, J.K. Whole genome sequencing and de novo assembly identifies Sydney-like variant noroviruses and recombinants during the winter 2012/2013 outbreak in England. Virol. J. 2013, 10, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mans, J.; Murray, T.Y.; Taylor, M.B. Novel norovirus recombinants detected in South Africa. Virol. J. 2014, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Koo, E.S.; Kim, M.S.; Choi, J.D.; Shin, Y.; Jeong, Y.S. Re-emergence of a GII. 4 norovirus Sydney 2012 variant equipped with GII. P16 RdRp and its predominance over novel variants of GII. 17 in South Korea in 2016. Food Environ. Virol. 2017, 9, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Shimizu, T.; Ishikawa, M.; Komane, A.; Okabe, N.; Ryo, A.; Kimura, H.; Katayama, K.; Shimizu, H. Complete genome sequence of a recombinant GII. P16-GII. 4 norovirus detected in Kawasaki City, Japan, in 2016. Genome Announc. 2016, 4, e01016–e01099. [Google Scholar] [CrossRef] [PubMed]
- Niendorf, S.; Jacobsen, S.; Faber, M.; Eis-Hübinger, A.; Hofmann, J.; Zimmermann, O.; Höhne, M.; Bock, C. Steep rise in norovirus cases and emergence of a new recombinant strain GII. P16-GII. 2, Germany, winter 2016. Euro Surveill. 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Matsushima, Y.; Motoya, T.; Mizukoshi, F.; Ueki, Y.; Sakon, N.; Murakami, K.; Shimizu, T.; Okabe, N.; Nagata, N. Phylogeny and immunoreactivity of Norovirus GII. P16-GII. 2, Japan, winter 2016–17. Emerg. Infect. Dis. 2018, 24, 144. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Barclay, L.; Collins, N.R.; Wikswo, M.E.; Castro, C.J.; Magaña, L.C.; Gregoricus, N.; Marine, R.L.; Chhabra, P.; Vinjé, J. Genetic and epidemiologic trends of norovirus outbreaks in the US demonstrated emergence of novel GII. 4 recombinant viruses, 2013–2016. J. Clin. Microbiol. 2017, 55, 2208–2221. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.; Wang, J.; Ling, H.; He, Y.; Dong, X.; Wang, X.; Peng, J.; Zhang, H.; Jin, M.; Duan, Z. Norovirus GII. P16/GII. 2–associated gastroenteritis, China, 2016. Emerg. Infect. Dis. 2017, 23, 1172. [Google Scholar] [CrossRef] [PubMed]
- Bidalot, M.; Théry, L.; Kaplon, J.; De Rougemont, A.; Ambert-Balay, K. Emergence of new recombinant noroviruses GII. P16-GII. 4 and GII. P16-GII. 2, France, winter 2016 to 2017. Euro Surveill. 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Greening, G.E.; Hewitt, J.; Rivera-Aban, M.; Croucher, D. Molecular epidemiology of norovirus gastroenteritis outbreaks in New Zealand from 2002–2009. J. Med. Virol. 2012, 84, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Hewitt, J.; Sitabkhan, A.; Eden, J.-S.; Lun, J.; Levy, A.; Merif, J.; Smith, D.; Rawlinson, W.D.; White, P.A. A multi-site study of norovirus molecular epidemiology in Australia and New Zealand, 2013–2014. PLoS ONE 2016, 11, e0145254. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Maust, B.S.; Nickle, D.C.; Learn, G.H.; Liu, Y.; Heath, L.; Pond, S.L.K.; Mullins, J.I. DIVEIN: A web server to analyze phylogenies, sequence divergence, diversity, and informative sites. Biotechniques 2010, 48, 405. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Bruggink, L.; Witlox, K.; Huang, B.; Warrilow, D.; Marshall, J. GII. 4 norovirus recombinant causes gastroenteritis epidemic in Eastern Australia, winter 2017. J. Med. Virol. 2018, 90, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Kazama, S.; Masago, Y.; Tohma, K.; Souma, N.; Imagawa, T.; Suzuki, A.; Liu, X.; Saito, M.; Oshitani, H.; Omura, T. Temporal dynamics of norovirus determined through monitoring of municipal wastewater by pyrosequencing and virological surveillance of gastroenteritis cases. Water Res. 2016, 92, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beek, J.; Ambert-Balay, K.; Botteldoorn, N.; Eden, J.; Fonager, J.; Hewitt, J.; Iritani, N.; Kroneman, A.; Vennema, H.; Vinje, J. Indications for worldwide increased norovirus activity associated with emergence of a new variant of genotype II. 4, late 2012. Euro Surveill. 2013, 18, 8–9. [Google Scholar] [PubMed]
- Farsi, M.; Roodbari, F.; Nejati, B.; Arashkia, A.; Jalilvand, S.; Nateghian, A.; Rahbarimanesh, A.; Marashi, S.M.; Shoja, Z. Prevalence and genetic diversity of norovirus genogroup II in children less than 5 years of age with acute gastroenteritis in Tehran, Iran. Med. Microbiol. Immunol. 2018, 207, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Ruis, C.; Roy, S.; Brown, J.R.; Allen, D.J.; Goldstein, R.A.; Breuer, J. The emerging GII. P16-GII. 4 Sydney 2012 norovirus lineage is circulating worldwide, arose by late-2014 and contains polymerase changes that may increase virus transmission. PLoS ONE 2017, 12, e0179572. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.; Sosnovtsev, S.V.; Green, K.Y. Static and evolving norovirus genotypes: Implications for epidemiology and immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic analyses suggest that factors other than the capsid protein play a role in the epidemic potential of GII. 2 norovirus. mSphere 2017, 2, e00117–e00187. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Eden, J.-S.; Rawlinson, W.D.; White, P.A. Rapid evolution of pandemic noroviruses of the GII. 4 lineage. PLoS Pathog. 2010, 6, e1000831. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Brewer-Jensen, P.D.; Mallory, M.L.; Debbink, K.; Swann, E.W.; Vinjé, J.; Baric, R.S. Antigenic characterization of a novel recombinant GII. P16-GII. 4 Sydney norovirus strain with minor sequence variation leading to antibody escape. J. Infect. Dis. 2017, 217, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Walker, C.M. Mutational escape from CD8+ T cell immunity: HCV evolution, from chimpanzees to man. J. Exp. Med. 2005, 201, 1709–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.W.; Shen, L.; Miller, M.D.; Ghim, S.H.; Hughes, A.L.; Letvin, N.L. Cytotoxic T lymphocytes do not appear to select for mutations in an immunodominant epitope of simian immunodeficiency virus gag. J. Immunol. 1992, 149, 4060–4066. [Google Scholar] [PubMed]
- Lindesmith, L.C.; Beltramello, M.; Donaldson, E.F.; Corti, D.; Swanstrom, J.; Debbink, K.; Lanzavecchia, A.; Baric, R.S. Immunogenetic mechanisms driving norovirus GII. 4 antigenic variation. PLoS Pathog. 2012, 8, e1002705. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Costantini, V.; Beltramello, M.; Corti, D.; Swanstrom, J.; Lanzavecchia, A.; Vinjé, J.; Baric, R.S. Emergence of new pandemic GII. 4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 2013, 208, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Lindesmith, L.C.; LoBue, A.D.; Baric, R.S. Viral shape-shifting: Norovirus evasion of the human immune system. Nat. Rev. Microbiol. 2010, 8, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Laskey, S.; Wasilewski, L.; Munshaw, S.; Fanning, L.J.; Kenny-Walsh, E.; Ray, S.C. Constraints on viral evolution during chronic hepatitis C virus infection arising from a common-source exposure. J. Virol. 2012, 86, 12582–12590. [Google Scholar] [CrossRef] [PubMed]
- Botosso, V.F.; Paolo, M.d.A.; Ueda, M.; Arruda, E.; Gilio, A.E.; Vieira, S.E.; Stewien, K.E.; Peret, T.C.; Jamal, L.F.; de MC Pardini, M.I. Positive selection results in frequent reversible amino acid replacements in the G protein gene of human respiratory syncytial virus. PLoS Pathog. 2009, 5, e1000254. [Google Scholar] [CrossRef] [PubMed]
- Fryer, H.R.; Frater, J.; Duda, A.; Roberts, M.G.; Phillips, R.E.; McLean, A.R.; SPARTAC Trial Investigators. Modelling the evolution and spread of HIV immune escape mutants. PLoS Pathog. 2010, 6, e1001196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A. A genotype network reveals homoplastic cycles of convergent evolution in influenza A (H3N2) haemagglutinin. Proc. Biol. Sci. 2014, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.H.; Sanders, L.P.; Tanaka, M.M. Modelling the role of immunity in reversion of viral antigenic sites. J. Theor. Biol. 2016, 392, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wikramaratna, P.S.; Sandeman, M.; Recker, M.; Gupta, S. The antigenic evolution of influenza: Drift or thrift? Phil. Trans. R. Soc. B 2013, 368, 20120200. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lun, J.H.; Hewitt, J.; Yan, G.J.H.; Enosi Tuipulotu, D.; Rawlinson, W.D.; White, P.A. Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017. Viruses 2018, 10, 548. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100548
Lun JH, Hewitt J, Yan GJH, Enosi Tuipulotu D, Rawlinson WD, White PA. Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017. Viruses. 2018; 10(10):548. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100548
Chicago/Turabian StyleLun, Jennifer H., Joanne Hewitt, Grace J. H. Yan, Daniel Enosi Tuipulotu, William D. Rawlinson, and Peter A. White. 2018. "Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017" Viruses 10, no. 10: 548. https://0-doi-org.brum.beds.ac.uk/10.3390/v10100548