Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Sclerotinia sclerotiorum Cultures and RNA Extraction
2.2. Analysis of S. sclerotiorum Transcriptome
2.3. Analysis of S. sclerotiorum Small RNA Populations
2.4. High-Throughput RNA Ligase-Mediated Rapid Amplification of cDNA Ends (HT-RACE) Analysis
3. Results
3.1. Changes in mRNA Accumulation Associated with Infection of S. sclerotiorum by SsHV2-L
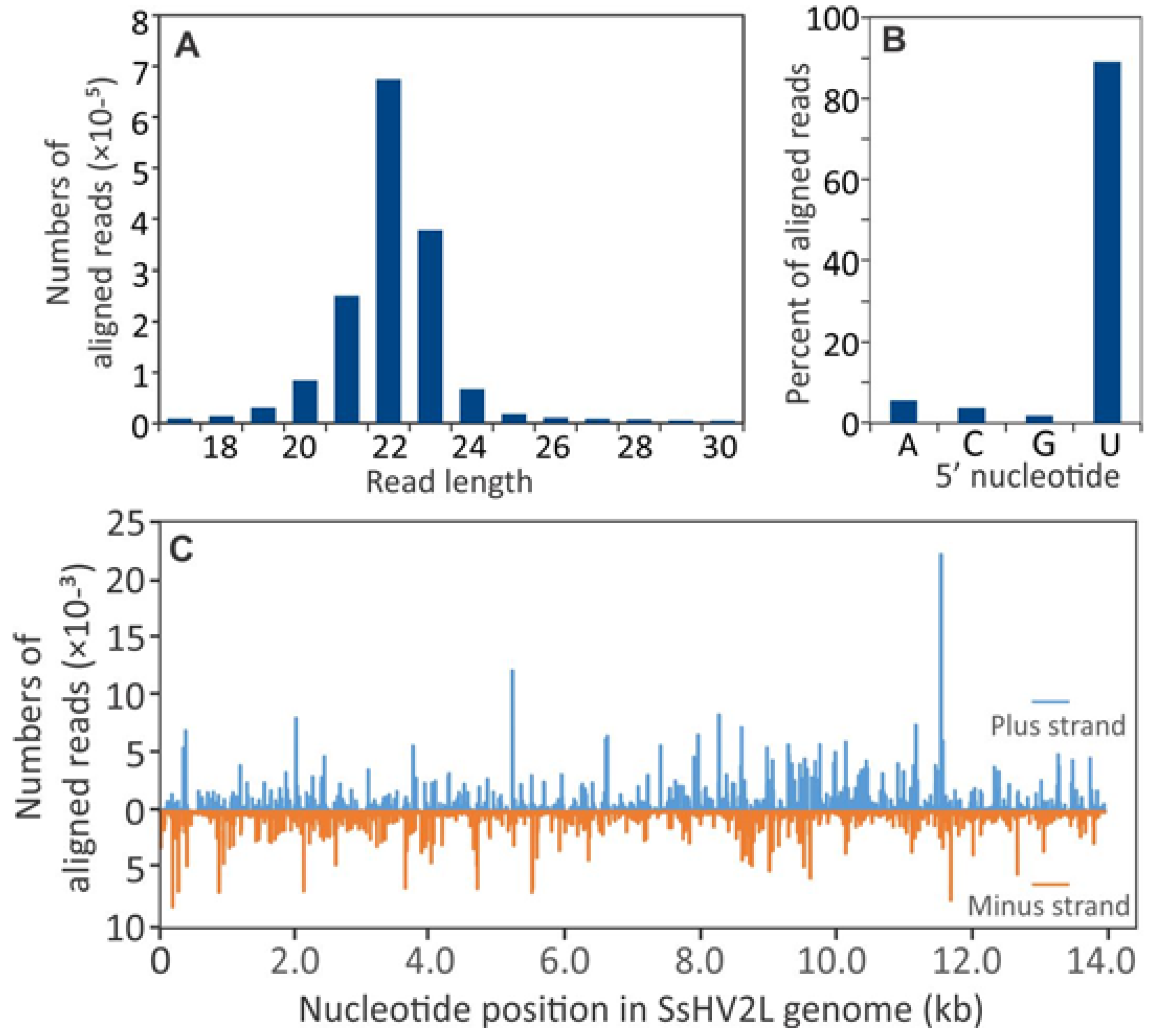
3.2. Small RNA Accumulation in Healthy and SsHV2-L-infected S. sclerotiorum Cultures
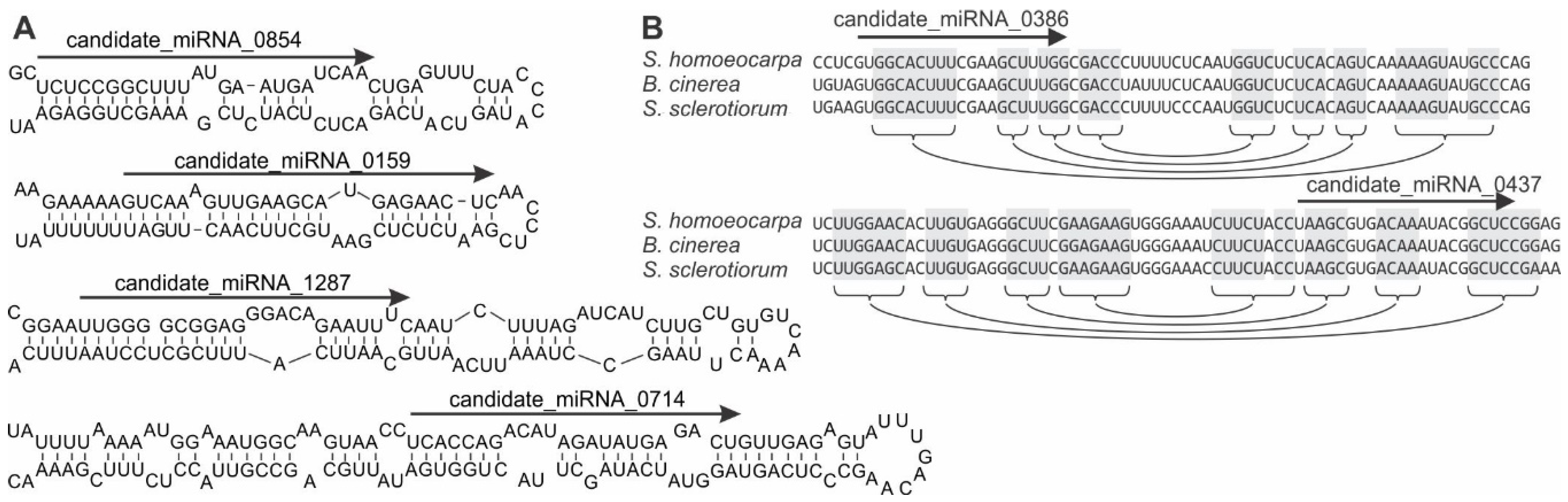
3.3. Identification of S. sclerotiorum Loci Producing microRNA-Like RNAs
3.4. Potential Targets for S. sclerotiorum sRNAs
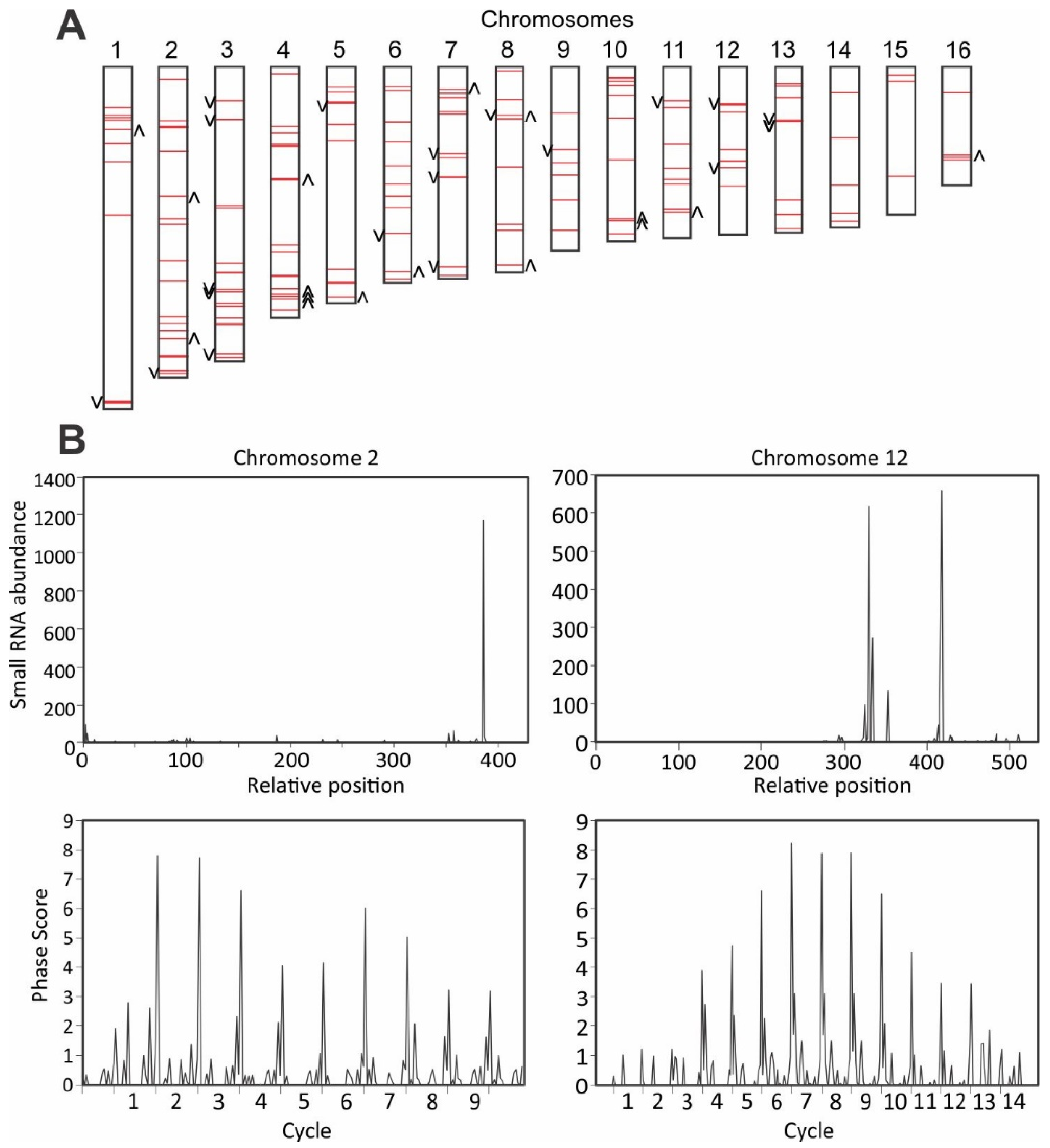
3.5. Detection of Phased siRNAs from S. sclerotiorum Noncoding RNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ghabrial, S.A.; Castón, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479–480, 356–368. [Google Scholar] [CrossRef]
- Nuss, D.L. Mycoviruses, RNA silencing, and viral RNA recombination. Adv. Virus Res. 2011, 80, 25–48. [Google Scholar]
- Baulcombe, D. RNA silencing. Trends Biochem. Sci. 2005, 30, 290–293. [Google Scholar] [CrossRef]
- Waterhouse, P.M.; Wang, M.B.; Lough, T. Gene silencing as an adaptive defence against viruses. Nature 2001, 411, 834–842. [Google Scholar] [CrossRef]
- Li, Y.; Lu, J.F.; Han, Y.H.; Fan, X.X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013, 342, 231–234. [Google Scholar] [CrossRef]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef]
- Nakayashiki, H.; Kadotani, N.; Mayama, S. Evolution and diversification of RNA silencing proteins in fungi. J. Mol. Evol. 2006, 63, 127–135. [Google Scholar] [CrossRef]
- Laurie, J.D.; Ali, S.; Linning, R.; Mannhaupt, G.; Wong, P.; Guldener, U.; Munsterkotter, M.; Moore, R.; Kahmann, R.; Bakkeren, G.; et al. Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species-specific presence of transposable elements. Plant Cell 2012, 24, 1733–1745. [Google Scholar] [CrossRef]
- Bernstein, D.A.; Vyas, V.K.; Weinberg, D.E.; Drinnenberg, I.A.; Bartel, D.P.; Fink, G.R. Candida albicans Dicer (CaDcr1) is required for efficient ribosomal and spliceosomal RNA maturation. Proc. Natl. Acad. Sci. USA 2012, 109, 523–528. [Google Scholar] [CrossRef]
- Axtell, M.J.; Westholm, J.O.; Lai, E.C. Vive la différence: Biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011, 12, 221. [Google Scholar] [CrossRef]
- Song, X.W.; Li, P.C.; Zhai, J.X.; Zhou, M.; Ma, L.J.; Liu, B.; Jeong, D.H.; Nakano, M.; Cao, S.Y.; Liu, C.Y.; et al. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012, 69, 462–474. [Google Scholar] [CrossRef]
- Arikit, S.; Zhai, J.X.; Meyers, B.C. Biogenesis and function of rice small RNAs from non-coding RNA precursors. Curr. Opin. Plant Biol. 2013, 16, 170–179. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.X.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Mueth, N.A.; Ramachandran, S.R.; Hulbert, S.H. Small RNAs from the wheat stripe rust fungus (Puccinia striiformis f.sp. tritici). BMC Genom. 2015, 16, 718. [Google Scholar] [CrossRef]
- Lin, R.M.; He, L.Y.; He, J.Y.; Qin, P.G.; Wang, Y.R.; Deng, Q.M.; Yang, X.T.; Li, S.C.; Wang, S.Q.; Wang, W.M.; et al. Comprehensive analysis of microRNA-Seq and target mRNAs of rice sheath blight pathogen provides new insights into pathogenic regulatory mechanisms. DNA Res. 2016, 23, 415–425. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, N.; Jiang, Q.Y.; Sun, X.J.; Wang, Y.; Zhang, H.; Hu, Z. Exploring microRNA-like small RNAs in the filamentous fungus Fusarium oxysporum. PLoS ONE 2014, 9, e104956. [Google Scholar] [CrossRef]
- Bai, Y.H.; Lan, F.X.; Yang, W.Q.; Zhang, F.; Yang, K.L.; Li, Z.G.; Gao, P.L.; Wang, S.H. SRNA profiling in Aspergillus flavus reveals differentially expressed miRNA-like RNAs response to water activity and temperature. Fungal Genet. Biol. 2015, 81, 113–119. [Google Scholar] [CrossRef]
- Thompson, D.M.; Parker, R. Stressing out over tRNA cleavage. Cell 2009, 138, 215–219. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, M.H.; Cao, Z.H.; Li, X.; Zhang, Y.F.; Shi, J.C.; Feng, G.H.; Peng, H.Y.; Zhang, X.D.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef]
- Martinez, G.; Choudury, S.G.; Slotkin, R.K. tRNA-derived small RNAs target transposable element transcripts. Nucleic Acids Res. 2017, 45, 5142–5152. [Google Scholar] [CrossRef] [Green Version]
- Nunes, C.C.; Gowda, M.; Sailsbery, J.; Xue, M.; Chen, F.; Brown, D.E.; Oh, Y.; Mitchell, T.K.; Dean, R.A. Diverse and tissue-enriched small RNAs in the plant pathogenic fungus, Magnaporthe oryzae. BMC Genom. 2011, 12, 288. [Google Scholar] [CrossRef]
- Wang, M.B.; Bian, X.Y.; Wu, L.M.; Liu, L.X.; Smith, N.A.; Isenegger, D.; Wu, R.M.; Masuta, C.; Vance, V.B.; Watson, J.M.; et al. On the role of RNA silencing in the pathogenicity and evolution of viroids and viral satellites. Proc. Natl. Acad. Sci. USA 2004, 101, 3275–3280. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Bao, F.S.; Xie, Z. Small RNA deep sequencing reveals role for arabidopsis thaliana RNA-dependent RNA polymerases in viral siRNA biogenesis. PLoS ONE 2009, 4, e4971. [Google Scholar] [CrossRef]
- Shimura, H.; Pantaleo, V.; Ishihara, T.; Myojo, N.; Inaba, J.; Sueda, K.; Burgyan, J.; Masuta, C. A viral satellite RNA induces yellow symptoms on tobacco by targeting a gene involved in chlorophyll biosynthesis using the RNA silencing machinery. PLoS Pathog. 2011, 7, e1002021. [Google Scholar] [CrossRef]
- Smith, N.A.; Eamens, A.L.; Wang, M.B. Viral small interfering RNAs target host genes to mediate disease symptoms in plants. PLoS Pathog. 2011, 7, e1002022. [Google Scholar] [CrossRef]
- Cao, M.J.; Du, P.; Wang, X.B.; Yu, Y.Q.; Qiu, Y.H.; Li, W.X.; Gal-On, A.; Zhou, C.Y.; Li, Y.; Ding, S.W. Virus infection triggers widespread silencing of host genes by a distinct class of endogenous siRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 14613–14618. [Google Scholar] [CrossRef]
- McBride, R.C.; Boucher, N.; Park, D.S.; Turner, P.E.; Townsend, J.P. Yeast response to la virus indicates coadapted global gene expression during mycoviral infection. FEMS Yeast Res. 2013, 13, 162–179. [Google Scholar] [CrossRef]
- Allen, T.D.; Dawe, A.L.; Nuss, D.L. Use of cdna microarrays to monitor transcriptional responses of the chestnut blight fungus cryphonectria parasitica to infection by virulence-attenuating hypoviruses. Eukaryot. Cell 2003, 2, 1253–1265. [Google Scholar] [CrossRef]
- Wang, J.Z.; Shi, L.M.; He, X.P.; Lu, L.D.; Li, X.P.; Chen, B.S. Comparative secretome analysis reveals perturbation of host secretion pathways by a hypovirus. Sci. Rep. 2016, 6, 34308. [Google Scholar] [CrossRef]
- Kwon, S.J.; Cho, S.Y.; Lee, K.M.; Yu, J.; Son, M.; Kim, K.H. Proteomic analysis of fungal host factors differentially expressed by Fusarium graminearum infected with fusarium graminearum virus-DK21. Virus Res. 2009, 144, 96–106. [Google Scholar] [CrossRef]
- Marzano, S.L.; Hobbs, H.A.; Nelson, B.D.; Hartman, G.L.; Eastburn, D.E.; McCoppin, N.K.; Domier, L.L. Transfection of Sclerotinia sclerotiorum with in vitro transcripts of a naturally occurring interspecific recombinant of Sclerotinia sclerotiorum hypovirus 2 significantly reduces virulence of the fungus. J. Virol. 2015, 89, 5060–5071. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Braker1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mi, H.Y.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Mackowiak, S.D. Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr. Protoc. Bioinform. 2011, 36, 12.10.1–12.10.15. [Google Scholar]
- An, J.Y.; Lai, J.; Lehman, M.L.; Nelson, C.C. miRDeep*: An integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res. 2013, 41, 727–737. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W. Prediction of plant miRNA genes. In Plant MicroRNAs: Methods and Protocols; Humana Press: New York, NY, USA, 2010; pp. 19–30. [Google Scholar]
- An, J.Y.; Lai, J.; Sajjanhar, A.; Lehman, M.L.; Nelson, C.C. miRPlant: An integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinform. 2014, 15, 275. [Google Scholar] [CrossRef]
- Axtell, M.J. Shortstack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.R.; Yeoh, J.M.; Coruh, C.; Axtell, M.J. Improved placement of multi-mapping small RNAs. G3 2016, 6, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fahlgren, N.; Chapman, E.J.; Cumbie, J.S.; Sullivan, C.M.; Givan, S.A.; Kasschau, K.D.; Carrington, J.C. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell 2007, 19, 926–942. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Baker, B. Preparation of cDNA Library for dRNA-Seq. Bio-protocol 2012, 2, e302. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. Cleaveland: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef]
- Whitham, S.A.; Yang, C.L.; Goodin, M.M. Global impact: Elucidating plant responses to viral infection. Mol. Plant-Microbe Interact. 2006, 19, 1207–1215. [Google Scholar] [CrossRef]
- Spain, B.H.; Koo, D.; Ramakrishnan, M.; Dzudzor, B.; Colicelli, J. Truncated forms of a novel yeast protein suppress the lethality of a G-protein α subunit deficiency by interacting with the β subunit. J. Biol. Chem. 1995, 270, 25435–25444. [Google Scholar] [CrossRef]
- Wei, W. Transcriptomic Characterization of Soybean–Sclerotinia Sclerotiorum Interaction at Early Infection Stages. Ph.D. Thesis, University of Illinois at Urbana–Champaign, Champaign, IL, USA, 2017. [Google Scholar]
- Fei, Q.; Yu, Y.; Liu, L.; Zhang, Y.; Baldrich, P.; Dai, Q.; Chen, X.; Meyers, B.C. Biogenesis of a 22-nt microRNA in Phaseoleae species by precursor-programmed uridylation. Proc. Natl. Acad. Sci. USA 2018, 115, 8037–8042. [Google Scholar] [CrossRef]
- Hammond, T.M.; Spollen, W.G.; Decker, L.M.; Blake, S.M.; Springer, G.K.; Shiu, P.K.T. Identification of small RNAs associated with meiotic silencing by unpaired DNA. Genetics 2013, 194, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, G.; Song, N.; Wang, J.; Cao, L.; Jiang, H.; Ding, T. Complete mitochondrial genome sequence of the phytopathogenic fungi Sclerotinia sclerotiorum JX-21. Mitochondrial DNA Part B 2016, 1, 656–657. [Google Scholar] [CrossRef]
- Zhu, R.S.; Li, X.; Chen, Q.S. Discovering numerical laws of plant microRNA by evolution. Biochem. Biophys. Res. Commun. 2011, 415, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Bollmann, S.R.; Kasschau, K.D.; Cuperus, J.T.; Press, C.M.; Sullivan, C.M.; Chapman, E.J.; Hoyer, J.S.; Gilbert, K.B.; Grunwald, N.J.; et al. Phytophthora have distinct endogenous small RNA populations that include short interfering and microRNAs. PLoS ONE 2013, 8, e77181. [Google Scholar] [CrossRef] [PubMed]
- Pinzon, N.; Li, B.; Martinez, L.; Sergeeva, A.; Presumey, J.; Apparailly, F.; Seitz, H. microRNA target prediction programs predict many false positives. Genome Res. 2017, 27, 234–245. [Google Scholar] [CrossRef]
- Vaucheret, H.; Vazquez, F.; Crete, P.; Bartel, D.P. The action of argonaute1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Gene Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Dawe, A.L.; Van Voorhies, W.A.; Lau, T.A.; Ulanov, A.V.; Li, Z. Major impacts on the primary metabolism of the plant pathogen Cryphonectria parasitica by the virulence-attenuating virus CHV1-EP713. Microbiology 2009, 155, 3913–3921. [Google Scholar] [CrossRef]
- Wang, S.C.; Zhang, J.Z.; Li, P.F.; Qiu, D.W.; Guo, L.H. Transcriptome-based discovery of Fusarium graminearum stress responses to FgHV1 infection. Int. J. Mol. Sci. 2016, 17, 1922. [Google Scholar] [CrossRef]
- Meena, M.; Prasad, V.; Zehra, A.; Gupta, V.K.; Upadhyay, R.S. Mannitol metabolism during pathogenic fungal-host interactions under stressed conditions. Front. Microbiol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef]
- Laluk, K.; AbuQamar, S.; Mengiste, T. The Arabidopsis mitochondria-localized pentatricopeptide repeat protein PGN functions in defense against necrotrophic fungi and abiotic stress tolerance. Plant Physiol. 2011, 156, 2053–2068. [Google Scholar] [CrossRef]
- Zhu, M.K.; Chen, G.P.; Dong, T.T.; Wang, L.L.; Zhang, J.L.; Zhao, Z.P.; Hu, Z.L. SLDEAD31, a putative DEAD-box RNA helicase gene, regulates salt and drought tolerance and stress-related genes in tomato. PLoS ONE 2015, 10, e0133849. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ge, L.L.; Li, P.P.; Wang, Y.; Sun, M.X.; Huang, L.; Ishag, H.; Di, D.D.; Shen, Z.Q.; Fan, W.X.; et al. The DEAD-box RNA helicase DDX5 acts as a positive regulator of Japanese encephalitis virus replication by binding to viral 3′ UTR. Antivir. Res. 2013, 100, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, N.; Pogany, J.; Nagy, P.D. A co-opted dead-box RNA helicase enhances tombusvirus plus-strand synthesis. PLoS Pathog. 2012, 8, e1002537. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Suzuki, N. Highly activated RNA silencing via strong induction of dicer by one virus can interfere with the replication of an unrelated virus. Proc. Natl. Acad. Sci. USA 2015, 112, E4911–E4918. [Google Scholar] [CrossRef]
- Zhang, X.M.; Segers, G.C.; Sun, Q.H.; Deng, F.Y.; Nuss, D.L. Characterization of hypovirus-derived small RNAs generated in the chestnut blight fungus by an inducible DCL-2-dependent pathway. J. Virol. 2008, 82, 2613–2619. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Choi, G.H.; Nuss, D.L. A single argonaute gene is required for induction of RNA silencing antiviral defense and promotes viral RNA recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 17927–17932. [Google Scholar] [CrossRef] [PubMed]
- Yaegashi, H.; Shimizu, T.; Ito, T.; Kanematsu, S. Differential inductions of RNA silencing among encapsidated double-stranded RNA mycoviruses in the white root rot fungus Rosellinia necatrix. J. Virol. 2016, 90, 5677–5692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.H.; Fu, Y.P.; Xie, J.T.; Li, B.; Jiang, D.H.; Li, G.Q.; Cheng, J.S. Identification of microRNA-like RNAs in a plant pathogenic fungus Sclerotinia sclerotiorum by high-throughput sequencing. Mol. Genet. Genom. 2012, 287, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Chow, W.N.; Wong, A.Y.P.; Yeung, J.M.Y.; Bao, J.; Zhang, N.; Lok, S.; Woo, P.C.Y.; Yuen, K.Y. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei. PLoS Negl. Trop. Dis. 2013, 7, e2398. [Google Scholar] [CrossRef]
- Kang, K.; Zhong, J.S.; Jiang, L.; Liu, G.; Gou, C.Y.; Wu, Q.; Wang, Y.; Luo, J.; Gou, D.M. Identification of microRNA-like RNAs in the filamentous fungus Trichoderma reesei by Solexa sequencing. PLoS ONE 2013, 8, e76288. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, T.A.; Kuck, U. Dicer-dependent biogenesis of small RNAs and evidence for microRNA-like RNAs in the penicillin producing fungus Penicillium chrysogenum. PLoS ONE 2015, 10, e0125989. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Zhang, Z.Y.; Liu, Y. RNA interference pathways in fungi: Mechanisms and functions. Annu. Rev. Microbiol. 2012, 66, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Williamson, V.; Kim, A.; Xie, B.; McMichael, G.O.; Gao, Y.; Vladimirov, V. Detecting miRNAs in deep-sequencing data: A software performance comparison and evaluation. Brief. Bioinform. 2013, 14, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.M.; Hellens, R.P. Coding in non-coding RNAs. Nature 2015, 520, 41–42. [Google Scholar] [CrossRef]
- Wang, Q.H.; Li, T.T.; Xu, K.; Zhang, W.; Wang, X.L.; Quan, J.L.; Jin, W.B.; Zhang, M.X.; Fan, G.J.; Wang, M.B.; et al. The tRNA-derived small RNAs regulate gene expression through triggering sequence-specific degradation of target transcripts in the oomycete pathogen Phytophthora sojae. Front. Plant Sci. 2016, 7, 1938. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keam, S.P.; Hutvagner, G. tRNA-derived fragments (tRFs): Emerging new roles for an ancient RNA in the regulation of gene expression. Life 2015, 5, 1638–1651. [Google Scholar] [CrossRef] [PubMed]
- Komiya, R. Biogenesis of diverse plant phasiRNAs involves an miRNA-trigger and Dicer-processing. J. Plant Res. 2017, 130, 17–23. [Google Scholar] [CrossRef]
- Fei, Q.L.; Xia, R.; Meyers, B.C. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef]
- Da Silva, R.P.; Puccia, R.; Rodrigues, M.L.; Oliveira, D.L.; Joffe, L.S.; Cesar, G.V.; Nimrichter, L.; Goldenberg, S.; Alves, L.R. Extracellular vesicle-mediated export of fungal RNA. Sci. Rep. 2015, 5, 7763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Qiao, L.; Wang, M.; He, B.; Lin, F.-M.; Palmquist, J.; Huang, H.-D.; Jin, H. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 2018, 360, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.W.; Zhang, Z.H.; Kaloshian, I.; Huang, H.D.; Jin, H.L. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Chen, L.T.; Patel, K.; Li, Y.H.; Baulcombe, D.C.; Wu, S.H. 22-nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. USA 2010, 107, 15269–15274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochama, P.; Jadhav, P.; Neupane, A.; Marzano, S.-Y.L. Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum. Viruses 2018, 10, 214. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee Marzano, S.-Y.; Neupane, A.; Domier, L. Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus. Viruses 2018, 10, 713. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120713
Lee Marzano S-Y, Neupane A, Domier L. Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus. Viruses. 2018; 10(12):713. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120713
Chicago/Turabian StyleLee Marzano, Shin-Yi, Achal Neupane, and Leslie Domier. 2018. "Transcriptional and Small RNA Responses of the White Mold Fungus Sclerotinia sclerotiorum to Infection by a Virulence-Attenuating Hypovirus" Viruses 10, no. 12: 713. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120713