Phylodynamics of Influenza A/H1N1pdm09 in India Reveals Circulation Patterns and Increased Selection for Clade 6b Residues and Other High Mortality Mutants



Abstract
:1. Introduction
2. Materials and Methods
2.1. Compilation of Sequence Datasets
2.2. A/H1N1pdm09 Transmission within India
2.3. Predictors of Transmission within India
2.4. Positive Selection Analysis
3. Results
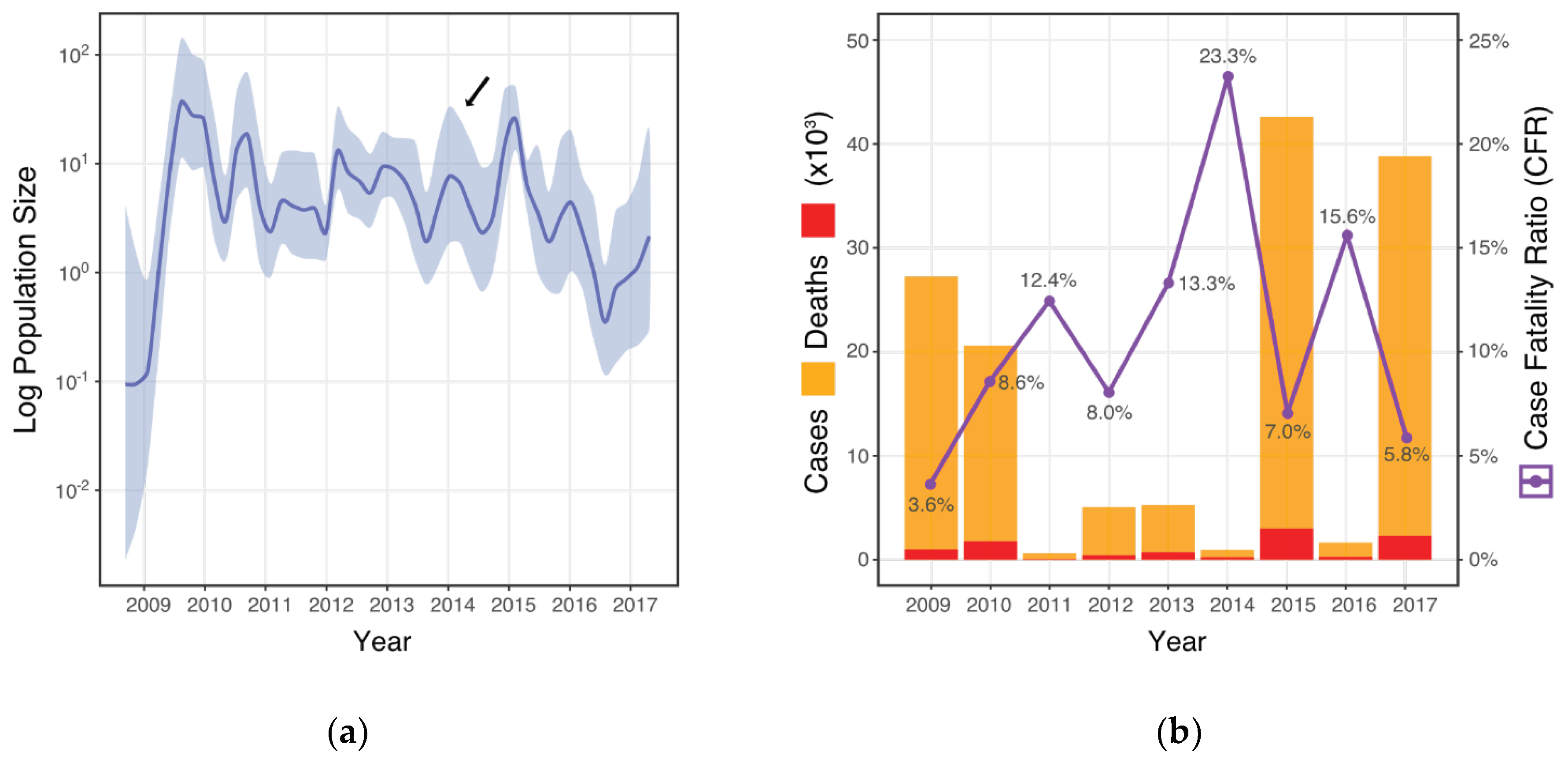
3.1. CFR and Viral Population Demographics
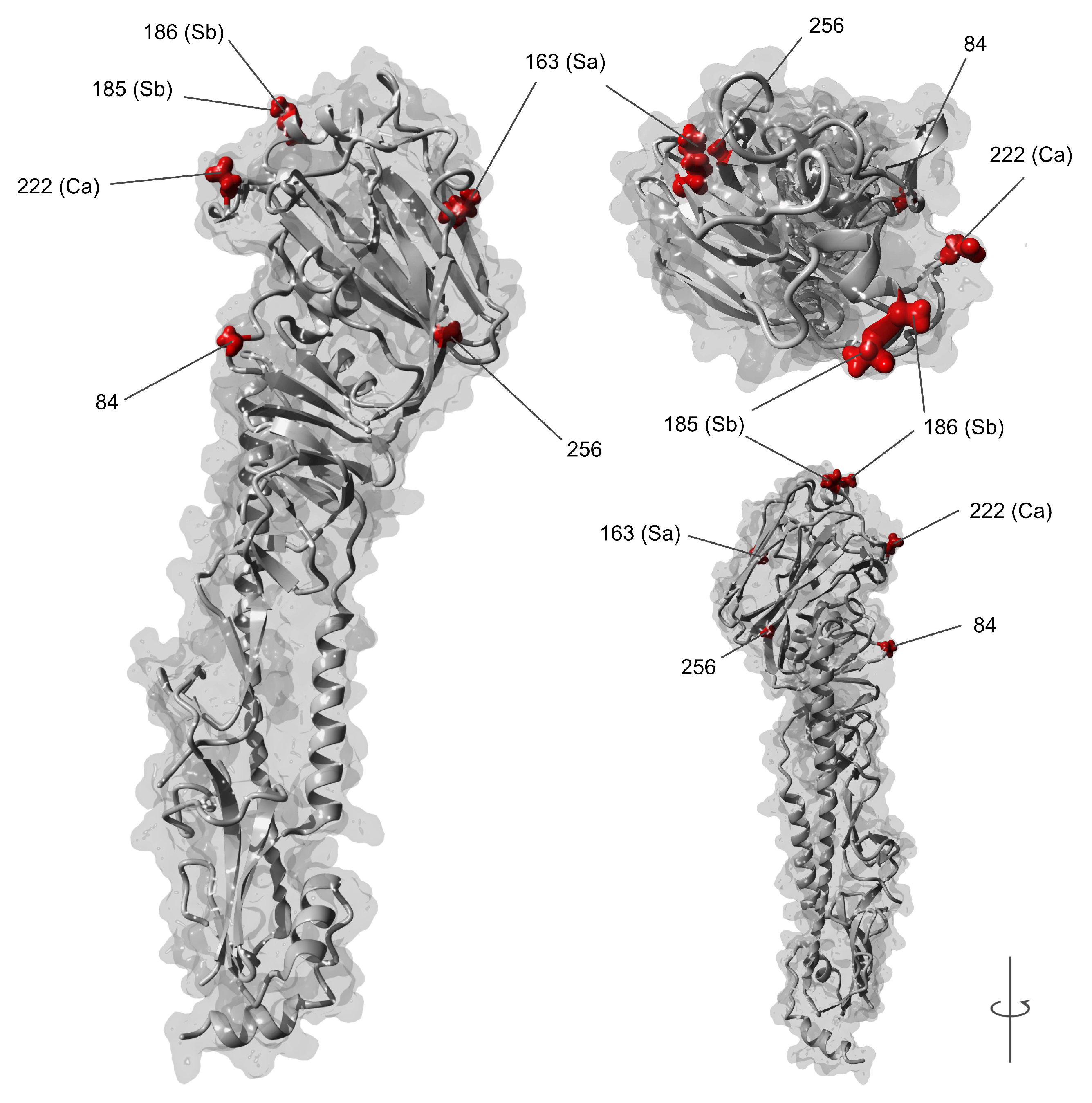
3.2. dN/dS Selection Analysis and Amino Acid Variations
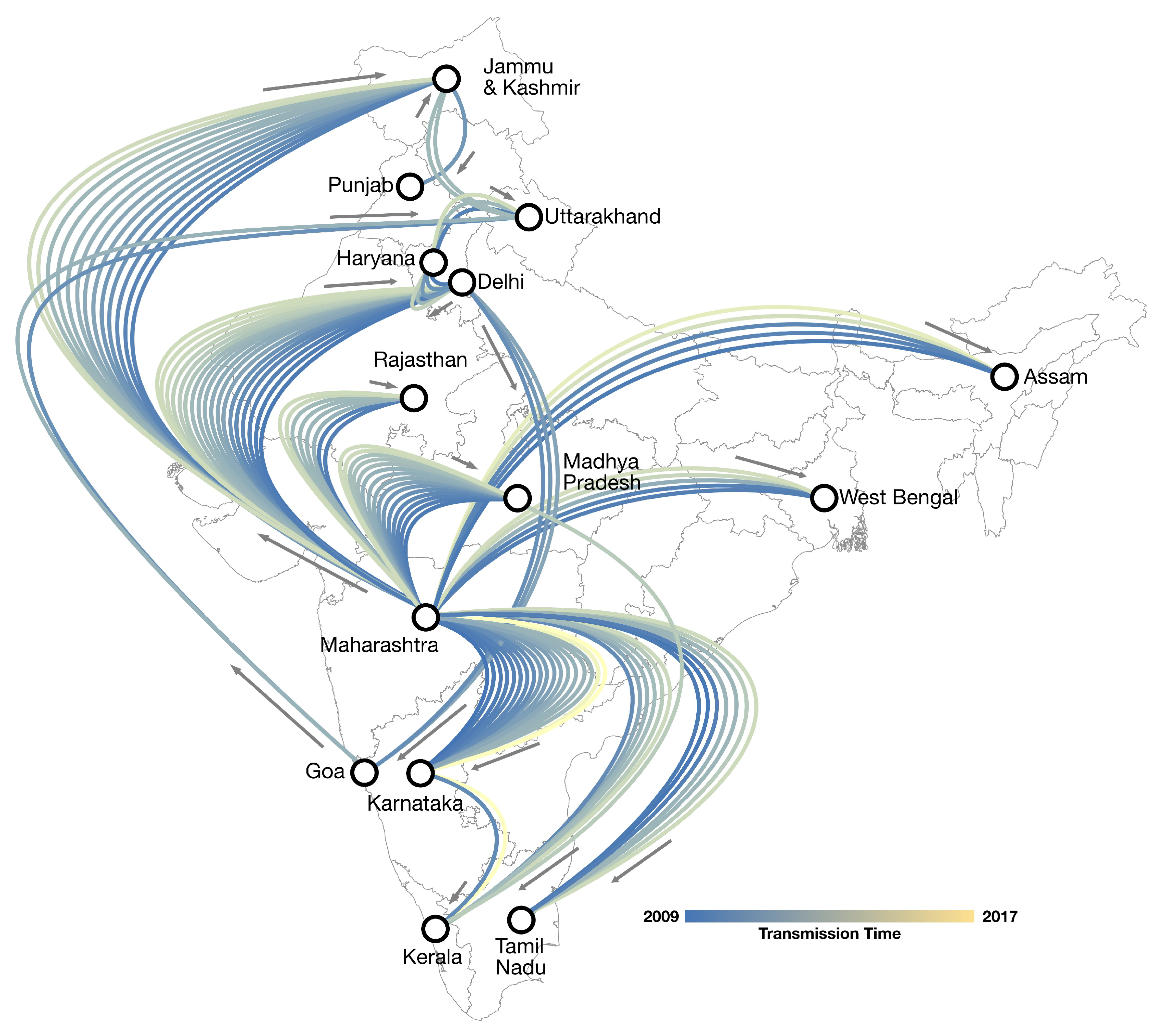
3.3. Phylogeography of A/H1N1pdm09
3.4. Generalized Linear Modelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Girard, M.P.; Tam, J.S.; Assossou, O.M.; Kieny, M.P. The 2009 A (H1N1) influenza virus pandemic: A review. Vaccine 2010, 28, 4895–4902. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Poon, L.L.M.; Guan, Y. Emergence of a novel swine-origin influenza a virus (S-OIV) H1N1 virus in humans. J. Clin. Virol. 2009, 45, 169–173. [Google Scholar] [CrossRef]
- Webb, S.A.R.; Pettilä, V.; Seppelt, I.; Bellomo, R.; Bailey, M.; Cooper, D.J.; Cretikos, M.; Davies, A.R.; Finfer, S.; Harrigan, P.W.J.; et al. Critical Care Services and 2009 H1N1 Influenza in Australia and New Zealand. N. Engl. J. Med. 2009, 361, 1925–1934. [Google Scholar] [PubMed] [Green Version]
- Mitchell, R.; Ogunremi, T.; Astrakianakis, G.; Bryce, E.; Gervais, R.; Gravel, D.; Johnston, L.; LeDuc, S.; Roth, V.; Taylor, G.; et al. Impact of the 2009 influenza A (H1N1) pandemic on Canadian health care workers: A survey on vaccination, illness, absenteeism, and personal protective equipment. Am. J. Infect. Control 2012, 40, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.Y.; Kelly, H.; Ip, D.K.M.; Wu, J.T.; Leung, G.M.; Cowling, B.J. Case fatality risk of influenza a (H1N1pdm09): A systematic review. Epidemiology 2013, 24, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Dawood, F.S.; Iuliano, A.D.; Reed, C.; Meltzer, M.I.; Shay, D.K.; Cheng, P.-Y.; Bandaranayake, D.; Breiman, R.F.; Brooks, W.A.; Buchy, P.; et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect. Dis. 2012, 12, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Van Kerkhove, M.D.; Hirve, S.; Koukounari, A.; Mounts, A.W. Estimating age-specific cumulative incidence for the 2009 influenza pandemic: a meta-analysis of A(H1N1)pdm09 serological studies from 19 countries. Influ. Other Respir. Viruses 2013, 7, 872–886. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, L.; Spreeuwenberg, P.; Lustig, R.; Taylor, R.J.; Fleming, D.M.; Kroneman, M.; Van Kerkhove, M.D.; Mounts, A.W.; Paget, W.J.; The GLaMOR Collaborating Teams. Global Mortality Estimates for the 2009 Influenza Pandemic from the GLaMOR Project: A Modeling Study. PLoS Med. 2013, 10, e1001558. [Google Scholar] [CrossRef]
- World Health Organization. H1N1 in Post-pandemic Period. Available online: https://www.who.int/mediacentre/news/statements/2010/h1n1_vpc_20100810/en/ (accessed on 31 December 2017).
- Broor, S.; Krishnan, A.; Roy, D.S.; Dhakad, S.; Kaushik, S.; Mir, M.A.; Singh, Y.; Moen, A.; Chadha, M.; Mishra, A.C.; et al. Dynamic Patterns of Circulating Seasonal and Pandemic A(H1N1)pdm09 Influenza Viruses From 2007–2010 in and around Delhi, India. PLOS ONE 2012, 7, e29129. [Google Scholar] [CrossRef]
- Bedford, T.; Riley, S.; Barr, I.G.; Broor, S.; Chadha, M.; Cox, N.J.; Daniels, R.S.; Gunasekaran, C.P.; Hurt, A.C.; Kelso, A.; et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 2015, 523, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Russell, C.A.; Jones, T.C.; Barr, I.G.; Cox, N.J.; Garten, R.J.; Gregory, V.; Gust, I.D.; Hampson, A.W.; Hay, A.J.; Hurt, A.C.; et al. The Global Circulation of Seasonal Influenza A (H3N2) Viruses. Science 2008, 320, 340–346. [Google Scholar] [CrossRef]
- Choudhry, A.; Singh, S.; Khare, S.; Rai, A.; Rawat, D.; Aggarwal, R.; Chauhan, L. Emergence of pandemic 2009 influenza A H1N1, India. Indian J. Med. Res. 2012, 135, 534–537. [Google Scholar]
- Gurav, Y.K.; Pawar, S.D.; Chadha, M.S.; A Potdar, V.; Deshpande, A.S.; Koratkar, S.S.; Hosmani, A.H.; Mishra, A.C. Pandemic influenza A(H1N1) 2009 outbreak in a residential school at Panchgani, Maharashtra, India. Indian J. Med. Res. 2010, 132, 67–71. [Google Scholar]
- Cases of Influenza a H1N1 (swine flu)—State/ut- Wise, Year- Wise for 2009, 2010, 2011 and 2012. Available online: http://mohfw. nic.in/showfile.php?lid=2121 (accessed on 31 December 2017).
- Seasonal Influenza (h1n1)– State/ut- Wise, Year- Wise Number of Cases and Death from 2010 to 2017. Available online: http://idsp.nic.in/showfile.php?lid=3908 (accessed on 31 December 2017).
- Murray, C.J.; Lopez, A.D.; Chin, B.; Feehan, D.; Hill, K.H. Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918–20 pandemic: A quantitative analysis. Lancet 2006, 368, 2211–2218. [Google Scholar] [CrossRef]
- Kumar, T.; Bhatia, D.; Lakshmi, P.V.M.; Laserson, K.F.; Narain, J.P. Risk factors for death during a resurgence of influenza-A (H1N1) pdm09 in Punjab State in 2013. Indian J. Public Health 2017, 61, 9. [Google Scholar] [CrossRef]
- Kulkarni, S.; Narain, J.; Gupta, S.; Dhariwal, A.; Singh, S.; MacIntyre, C. Influenza A (H1N1) in India: Changing epidemiology and its implications. Natl. Med. J. India 2019. [Google Scholar] [CrossRef]
- Malhotra, B.; Singh, R.; Sharma, P.; Meena, D.; Gupta, J.; Atreya, A.; Meena, B.R. Epidemiological & clinical profile of influenza A (H1N1) 2009 virus infections during 2015 epidemic in Rajasthan. Indian J. Med. Res. 2016, 144, 918–923. [Google Scholar]
- Baillie, G.J.; Galiano, M.; Agapow, P.-M.; Myers, R.; Chiam, R.; Gall, A.; Palser, A.L.; Watson, S.J.; Hedge, J.; Underwood, A. Evolutionary dynamics of local pandemic H1N1/2009 influenza virus lineages revealed by whole-genome analysis. J. Virol. 2012, 86, 11–18. [Google Scholar] [CrossRef]
- Hedge, J.; Lycett, S.J.; Rambaut, A. Real-time characterization of the molecular epidemiology of an influenza pandemic. Boil. Lett. 2013, 9, 20130331. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.R.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2. PLOS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Boil. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 16. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Boil. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate Model Selection of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol. Boil. Evol. 2012, 30, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, ew007. [Google Scholar] [CrossRef]
- Rambaut, A.; Suchard, M.; Xie, D.; Drummond, A. Tracer v1.6 (Software). Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 15 May 2019).
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth Skyride through a Rough Skyline: Bayesian Coalescent-Based Inference of Population Dynamics. Mol. Boil. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Boil. Evol. 2012, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Boil. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Boil. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, H. The Theory of Probability, 3rd ed.; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309. [Google Scholar] [CrossRef] [PubMed]
- Scotch, M.; Mei, C.; Makonnen, Y.J.; Pinto, J.; Ali, A.; Vegso, S.; Kane, M.; Sarkar, I.N.; Rabinowitz, P. Phylogeography of influenza A H5N1 clade 2.2.1.1 in Egypt. BMC Genom. 2013, 14, 871. [Google Scholar] [CrossRef] [PubMed]
- Domestic Traffic Reports. Available online: http://dgca.nic.in/reports/Traffic-ind.htm (accessed on 22 August 2019).
- Beard, R.; Magee, D.; Suchard, M.A.; Lemey, P.; Scotch, M. Generalized Linear Models for Identifying Predictors of the Evolutionary Diffusion of Viruses. AMIA Jt. Summits Transl. Sci. Proc. 2014, 2014, 23–28. [Google Scholar]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.K.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Boil. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Pond, S.L.K.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. In Practical Considerations for Adaptive Trial Design and Implementation; Springer Science and Business Media LLC: Berlin, Germany, 2005; pp. 125–181. [Google Scholar] [Green Version]
- Burke, D.F.; Smith, D.J. A Recommended Numbering Scheme for Influenza A HA Subtypes. PLOS ONE 2014, 9, e112302. [Google Scholar] [CrossRef]
- Yang, H.; Chang, J.C.; Guo, Z.; Carney, P.J.; Shore, D.A.; Donis, R.O.; Cox, N.J.; Villanueva, J.M.; Klimov, A.I.; Stevens, J. Structural Stability of Influenza A(H1N1)pdm09 Virus Hemagglutinins. J. Virol. 2014, 88, 4828–4838. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Nishiura, H. Case fatality ratio of pandemic influenza. Lancet Infect. Dis. 2010, 10, 443–444. [Google Scholar] [CrossRef]
- Cousins, S. Death toll from swine flu in India exceeds 2500. BMJ 2015, 351, 4966. [Google Scholar] [CrossRef]
- India Struggles with Deadly Swine Flu Outbreak. Available online: https://www.bbc.com/news/world-asia-india-31547455 (accessed on 18 January 2019).
- Swine Flu Deaths at 1895; Number of Cases near 32k Mark. Available online: https://indianexpress.com/article/india/india-others/swine-flu-deaths-at-1895-cases-near-32k-mark/ (accessed on 18 January 2019).
- Summary of the 2015–2016 Influenza Season. Available online: https://www.cdc.gov/flu/about/season/flu-season-2015–2016.htm (accessed on 23 January 2019).
- Influenza in Europe—Season 2013–2014; European Centre for Disease Prevention and Control (ECDC): Stockholm. Available online: https://ecdc.europa.eu/sites/portal/files/media/en/publications/Publications/Influenza-2013-14-season-report.pdf (accessed on 23 January 2019).
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47. [Google Scholar] [CrossRef]
- Treanor, J.J. 167—influenza (including avian influenza and swine influenza). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier Saunders: Philadelphia, Pennsylvania, 2015; pp. 2000–2024. [Google Scholar]
- Hay, A.J.; Gregory, V.; Douglas, A.R.; Lin, Y.P. The evolution of human influenza viruses. Philos. Trans. R. Soc. B Boil. Sci. 2001, 356, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, A.; Yamayoshi, S.; Soni, P.; Takenaga, T.; Kawakami, C.; Takashita, E.; Sakai-Tagawa, Y.; Uraki, R.; Ito, M.; Iwatsuki-Horimoto, K.; et al. Diversity of antigenic mutants of influenza A(H1N1)pdm09 virus escaped from human monoclonal antibodies. Sci. Rep. 2017, 7, 17735. [Google Scholar] [CrossRef]
- Horm, S.V.; Mardy, S.; Rith, S.; Ly, S.; Heng, S.; Vong, S.; Kitsutani, P.; Ieng, V.; Tarantola, A.; Ly, S. Epidemiological and virological characteristics of influenza viruses circulating in Cambodia from 2009 to 2011. PloS. ONE 2014, 9, e110713. [Google Scholar] [CrossRef]
- Nguyen, H.K.L.; Nguyen, P.T.K.; Nguyen, T.C.; Hoang, P.V.M.; Le, T.T.; Vuong, C.D.; Nguyen, A.P.; Tran, L.T.T.; Nguyen, B.G.; Le, M.Q. Virological characterization of influenza H1N1pdm09 in Vietnam, 2010-2013. Influ. Other Respir. Viruses 2015, 9, 216–224. [Google Scholar] [CrossRef]
- Pandey, S.; Sahu, M.; Potdar, V.; Barde, P. Molecular analysis of influenza A H1N1pdm09 virus circulating in Madhya Pradesh, India in the year 2017. VirusDisease 2018, 29, 380–384. [Google Scholar] [CrossRef]
- Ramos, A.P.; Herrera, B.A.; Ramírez, O.V.; García, A.A.; Jiménez, M.M.; Valdés, C.S.; Fernández, A.G.; González, G.; Fernández, S.I.O.; Báez, G.G.; et al. Molecular and phylogenetic analysis of influenza A H1N1 pandemic viruses in Cuba, May 2009 to August 2010. Int. J. Infect. Dis. 2013, 17, e565–e567. [Google Scholar] [CrossRef] [Green Version]
- Houng, H.-S.H.; Garner, J.; Zhou, Y.; Lyons, A.; Kuschner, R.; Deye, G.; Clair, K.S.; Douce, R.W.; Chicaiza, W.; Blair, P.J.; et al. Emergent 2009 influenza A(H1N1) viruses containing HA D222N mutation associated with severe clinical outcomes in the Americas. J. Clin. Virol. 2012, 53, 12–15. [Google Scholar] [CrossRef]
- Ledesma, J.; Pozo, F.; Reina, G.; Blasco, M.; Rodríguez, G.; Montes, M.; López-Miragaya, I.; Salvador, C.; Reina, J.; De Lejarazu, R.O.; et al. Genetic diversity of influenza A(H1N1)2009 virus circulating during the season 2010–2011 in Spain. J. Clin. Virol. 2012, 53, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, T.; De Rosa, F.; Cerutti, F.; Pagani, N.; Allice, T.; Stella, M.L.; Milia, M.G.; Calcagno, A.; Burdino, E.; Gregori, G.; et al. A(H1N1)pdm09 hemagglutinin D222G and D222N variants are frequently harbored by patients requiring extracorporeal membrane oxygenation and advanced respiratory assistance for severe A(H1N1)pdm09 infection. Influ. Other Respir. Viruses 2013, 7, 1416–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koel, B.F.; Burke, D.F.; Bestebroer, T.M.; Van Der Vliet, S.; Zondag, G.C.M.; Vervaet, G.; Skepner, E.; Lewis, N.S.; Spronken, M.I.J.; Russell, C.A.; et al. Substitutions Near the Receptor Binding Site Determine Major Antigenic Change During Influenza Virus Evolution. Science 2013, 342, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Llamas, R.; Alfaro-Ruiz, L.; Canon, C.A.; Rosshandler, I.I.; Cruz-Lagunas, A.; Zúñiga, J.; Vega, R.R.; Wong, C.W.; Maurer-Stroh, S.; Córdoba, S.R. Molecular features of influenza A (H1N1) pdm09 prevalent in Mexico during winter seasons 2012–2014. PloS. ONE 2017, 12, e0180419. [Google Scholar] [CrossRef] [PubMed]
- Parida, M.; Dash, P.K.; Kumar, J.S.; Joshi, G.; Tandel, K.; Sharma, S.; Srivastava, A.; Agarwal, A.; Saha, A.; Saraswat, S. Emergence of influenza A (H1N1) pdm09 Genogroup 6B and drug resistant virus, India, January to May 2015. Eurosurveillance 2016, 21, 30124. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Shirakura, M.; Fujisaki, S.; Kishida, N.; Burke, D.F.; Smith, D.J.; Kuwahara, T.; Takashita, E.; Takayama, I.; Nakauchi, M.; et al. Characterization of influenza A(H1N1)pdm09 viruses isolated from Nepalese and Indian outbreak patients in early 2015. Influ. Other Respir. Viruses 2017, 11, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Linderman, S.L.; Chambers, B.S.; Zost, S.J.; Parkhouse, K.; Li, Y.; Herrmann, C.; Ellebedy, A.H.; Carter, D.M.; Andrews, S.F.; Zheng, N.Y.; et al. Potential antigenic explanation for atypical H1N1 infections among middle-aged adults during the 2013–2014 influenza season. Proc. Natl. Acad. Sci. USA 2014, 111, 15798–15803. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Recommended composition of influenza virus vaccines for use in the 2017 southern hemisphere influenza. In Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2016; Volume 91. [Google Scholar]
- World Health Organization. Recommended composition of influenza virus vaccines for use in the 2019 southern hemisphere influenza. In Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2018; Volume 93, pp. 553–576. [Google Scholar]
- World Health Organization. Recommended composition of influenza virus vaccines for use in the 2019–2020 northern hemisphere influenza. In Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2019; Volume 94, pp. 141–160. [Google Scholar]
- Abed, Y.; Pizzorno, A.; Hamelin, M.-E.; Leung, A.; Joubert, P.; Couture, C.; Kobasa, D.; Boivin, G.; Pizzorno, M.A. The 2009 Pandemic H1N1 D222G Hemagglutinin Mutation Alters Receptor Specificity and Increases Virulence in Mice but Not in Ferrets. J. Infect. Dis. 2011, 204, 1008–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belser, J.A.; Jayaraman, A.; Raman, R.; Pappas, C.; Zeng, H.; Cox, N.J.; Katz, J.M.; Sasisekharan, R.; Tumpey, T.M. Effect of D222G Mutation in the Hemagglutinin Protein on Receptor Binding, Pathogenesis and Transmissibility of the 2009 Pandemic H1N1 Influenza Virus. PLOS ONE 2011, 6, e25091. [Google Scholar] [CrossRef] [PubMed]
- A Vazquez-Perez, J.; Isa, P.; Kobasa, D.; E Ormsby, C.; E Ramírez-Gonzalez, J.; Romero-Rodríguez, D.P.; Ranadheera, C.; Li, Y.; Bastien, N.; Embury-Hyatt, C.; et al. A (H1N1) pdm09 HA D222 variants associated with severity and mortality in patients during a second wave in Mexico. Virol. J. 2013, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Goka, E.A.; Vallely, P.J.; Mutton, K.J.; Klapper, P.E.; Goka, E. Mutations associated with severity of the pandemic influenza A(H1N1)pdm09 in humans: a systematic review and meta-analysis of epidemiological evidence. Arch. Virol. 2014, 159, 3167–3183. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Nayak, M.K.; Dutta, S.; Panda, S.; Satpathi, B.R.; Chawla-Sarkar, M. Genetic Characterization of Circulating 2015 A(H1N1)pdm09 Influenza Viruses from Eastern India. PLoS ONE 2016, 11, 0168464. [Google Scholar] [CrossRef] [PubMed]
- Tharakaraman, K.; Sasisekharan, R. Influenza Surveillance: 2014–2015 H1N1 “Swine”-Derived Influenza Viruses from India. Cell Host Microbe 2015, 17, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Press note 116939. Available online: http://pib.nic.in/newsite/printrelease.aspx?Relid=116939 (accessed on 2 May 2019).
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Taylor, J.; George, K.S.; Griesemer, S.B.; Ghedin, E.; Sengamalay, N.A.; Spiro, D.J.; et al. Stochastic Processes Are Key Determinants of Short-Term Evolution in Influenza A Virus. PLOS Pathog. 2006, 2, e125. [Google Scholar]
- Lowen, A.C.; Mubareka, S.; Steel, J.; Palese, P. Influenza Virus Transmission Is Dependent on Relative Humidity and Temperature. PLOS Pathog. 2007, 3, e151. [Google Scholar] [CrossRef] [PubMed]
- Lowen, A.C.; Steel, J. Roles of Humidity and Temperature in Shaping Influenza Seasonality. J. Virol. 2014, 88, 7692–7695. [Google Scholar] [CrossRef] [Green Version]
- Koul, P.A.; Broor, S.; Saha, S.; Barnes, J.; Smith, C.; Shaw, M.; Chadha, M.; Lal, R.B. Differences in Influenza Seasonality by Latitude, Northern India. Emerg. Infect. Dis. 2014, 20, 1746–1749. [Google Scholar] [CrossRef] [Green Version]
- Chadha, M.S.; Potdar, V.A.; Saha, S.; Koul, P.A.; Broor, S.; Dar, L.; Chawla-Sarkar, M.; Biswas, D.; Gunasekaran, P.; Abraham, A.M.; et al. Dynamics of Influenza Seasonality at Sub-Regional Levels in India and Implications for Vaccination Timing. PLOS ONE 2015, 10, 0124122. [Google Scholar] [CrossRef]
- Magee, D.; Suchard, M.A.; Scotch, M. Bayesian phylogeography of influenza A/H3N2 for the 2014-15 season in the United States using three frameworks of ancestral state reconstruction. PLoS Comput. Biol 2017, 13, 1005389. [Google Scholar] [CrossRef]
- De Maio, N.; Wu, C.-H.; O’Reilly, K.M.; Wilson, D. New Routes to Phylogeography: A Bayesian Structured Coalescent Approximation. PLoS Genet. 2015, 11, 1005421. [Google Scholar] [CrossRef]
- Indian Railways Statistical Publications 2016–17: Statistical summary—Indian Railways. Available online: http://www.indianrailways.gov.in/railwayboard/uploads/directorate/stat_econ/IRSP_2016–17/Annual_Report_Accounts_Eng/Statistical_Summary.pdf (accessed on 15 August 2019).
- Gao, Y.; Zhang, Y.; Shinya, K.; Deng, G.; Jiang, Y.; Li, Z.; Guan, Y.; Tian, G.; Li, Y.; Shi, J.; et al. Identification of Amino Acids in HA and PB2 Critical for the Transmission of H5N1 Avian Influenza Viruses in a Mammalian Host. PLOS Pathog. 2009, 5, e1000709. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Year | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S/UT (Population 106) | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | Total | % |
| Assam (31.2) | 4 | 4 | 1 | 11 | 20 | 3.3 | |||||
| Delhi (16.8) | 10 | 4 | 7 | 14 | 6 | 2 | 20 | 63 | 10.3 | ||
| Goa (1.5) | 1 | 3 | 3 | 3 | 10 | 1.6 | |||||
| Haryana (25.4) | 1 | 2 | 1 | 3 | 3 | 2 | 12 | 2.0 | |||
| Jammu and Kashmir (12.5) | 1 | 2 | 8 | 29 | 7 | 11 | 58 | 9.5 | |||
| Karnataka (61.1) | 19 | 16 | 5 | 15 | 6 | 61 | 10.0 | ||||
| Kerala (33.4) | 1 | 2 | 9 | 2 | 3 | 3 | 2 | 22 | 3.6 | ||
| Madhya Pradesh (72.6) | 4 | 8 | 4 | 5 | 5 | 18 | 44 | 7.2 | |||
| Maharashtra (112.4) | 42 | 8 | 5 | 54 | 66 | 6 | 53 | 8 | 242 | 39.5 | |
| Punjab (27.7) | 1 | 2 | 4 | 7 | 1.1 | ||||||
| Rajasthan (68.6) | 3 | 1 | 3 | 5 | 6 | 18 | 2.9 | ||||
| Tamil Nadu (72.15) | 5 | 2 | 2 | 6 | 1 | 16 | 2.6 | ||||
| Uttarakhand (10.1) | 1 | 1 | 1 | 2 | 1 | 6 | 1.0 | ||||
| West Bengal (91.3) | 3 | 5 | 26 | 34 | 5.5 | ||||||
| Year Total | 96 | 53 | 37 | 143 | 96 | 13 | 147 | 19 | 9 | 613 | 100.0 |
| Site (H3 a) | Ag b | India Taxa (n = 613) | International (S1) | International (S2) | |||
|---|---|---|---|---|---|---|---|
| dN/dS | 95% BCI | dN/dS | 95% BCI | dN/dS | 95% BCI | ||
| 84 (92) | n/a | 8.14 | (5.68–10.92) | 2.53 | (1.69–3.45) | 3.51 | (2.33–4.79) |
| 163 (166) | Sa | 3.83 | (2.68–5.35) | 4.54 | (2.98–6.38) | 2.62 | (1.79–3.68) |
| 185 (188) | Sb | 4.50 | (3.18–6.21) | 2.14 | (1.41–2.94) | 1.13 | (0.76–1.54) |
| 186 (189) | Sb | 3.35 | (2.26–4.51) | 1.44 | (0.97–1.94) | 1.47 | (0.93–1.97) |
| 222 (225) | Ca | 13.42 | (9.43–18.42) | 3.42 | (2.30–4.81) | 4.32 | (2.87–5.85) |
| 256 (259) | n/a | 4.39 | (3.12–6.09) | 1.16 | (0.74–1.58) | 1.08 | (0.75–1.48) |
| Predictor | E(δ) Probability | (β| δ) Coefficient | 95% BCI | BF a |
|---|---|---|---|---|
| Distance | 0.88 | −0.53 | −0.79 to −0.28 | 269.3 |
| SS origin | 0.87 | 1.46 | 0.96 to 2.12 | 237.2 |
| P Flux destination | 0.31 | 0.35 | 0.15 to 0.56 | 15.67 |
| SS destination | 0.22 | 0.26 | 0.11 to 0.42 | 10.03 |
| Pop Dense origin | 0.2 | −1.43 | −2.82 to −0.28 | 8.93 |
| P Flux origin | 0.14 | 1.86 | −0.46 to 2.95 | 5.84 |
| Average temp origin | 0.12 | −0.49 | −0.8 to −0.18 | 4.69 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adam, D.C.; Scotch, M.; MacIntyre, C.R. Phylodynamics of Influenza A/H1N1pdm09 in India Reveals Circulation Patterns and Increased Selection for Clade 6b Residues and Other High Mortality Mutants. Viruses 2019, 11, 791. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090791
Adam DC, Scotch M, MacIntyre CR. Phylodynamics of Influenza A/H1N1pdm09 in India Reveals Circulation Patterns and Increased Selection for Clade 6b Residues and Other High Mortality Mutants. Viruses. 2019; 11(9):791. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090791
Chicago/Turabian StyleAdam, Dillon C., Matthew Scotch, and C Raina. MacIntyre. 2019. "Phylodynamics of Influenza A/H1N1pdm09 in India Reveals Circulation Patterns and Increased Selection for Clade 6b Residues and Other High Mortality Mutants" Viruses 11, no. 9: 791. https://0-doi-org.brum.beds.ac.uk/10.3390/v11090791