Fatty Acids Regulate Porcine Reproductive and Respiratory Syndrome Virus Infection via the AMPK-ACC1 Signaling Pathway

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Reagents
2.2. Small Interfering RNAs (siRNAs) and Transfection
2.3. Cytotoxicity Assay
2.4. Western Blot Assay
2.5. Indirect Immunofluorescence Assay
2.6. TCID50 Assay
2.7. Plaque Assay
2.8. Statistical Analysis
2.9. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
3. Results
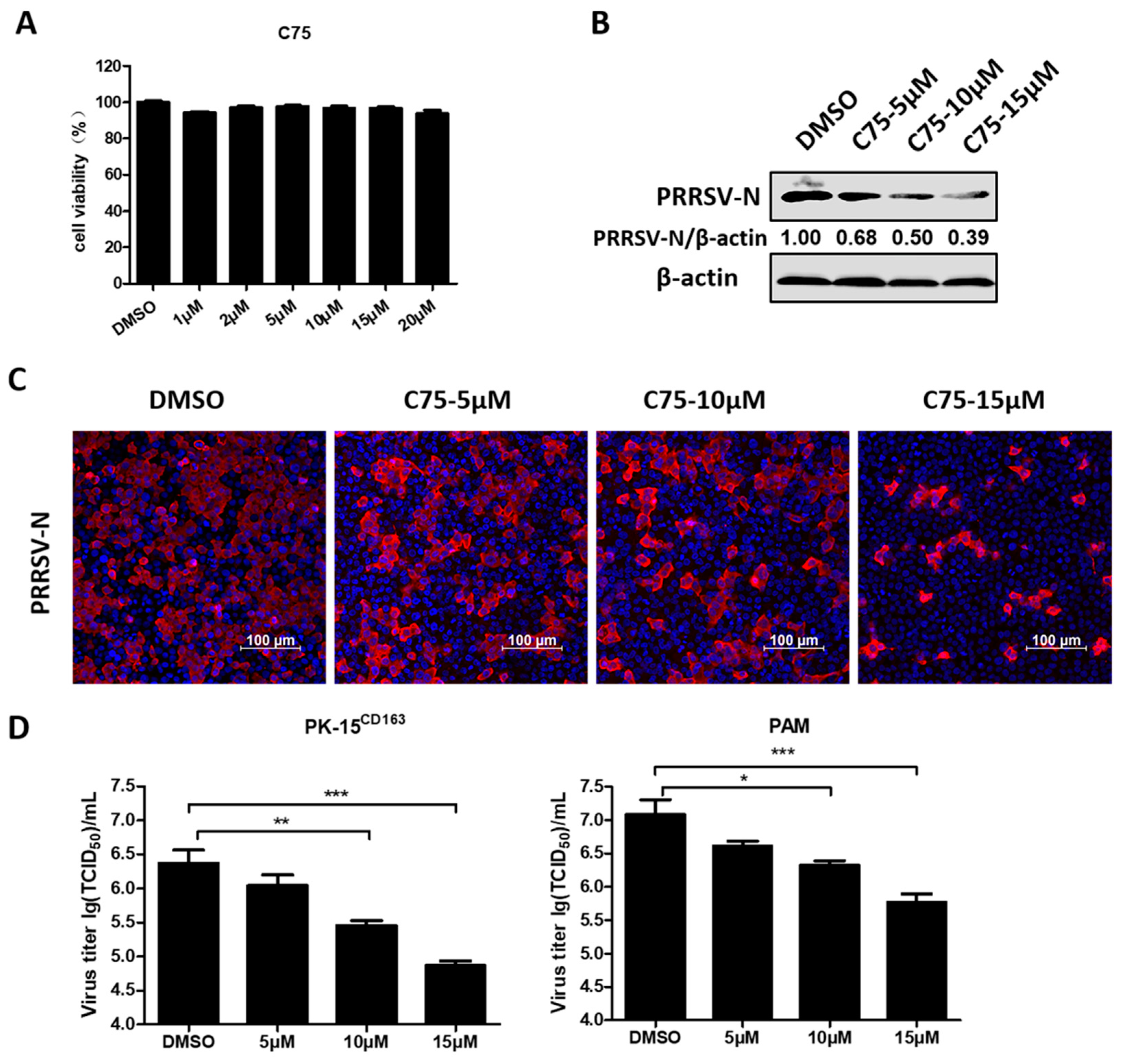
3.1. Blocking Fatty Acid Synthesis Inhibits PRRSV Replication
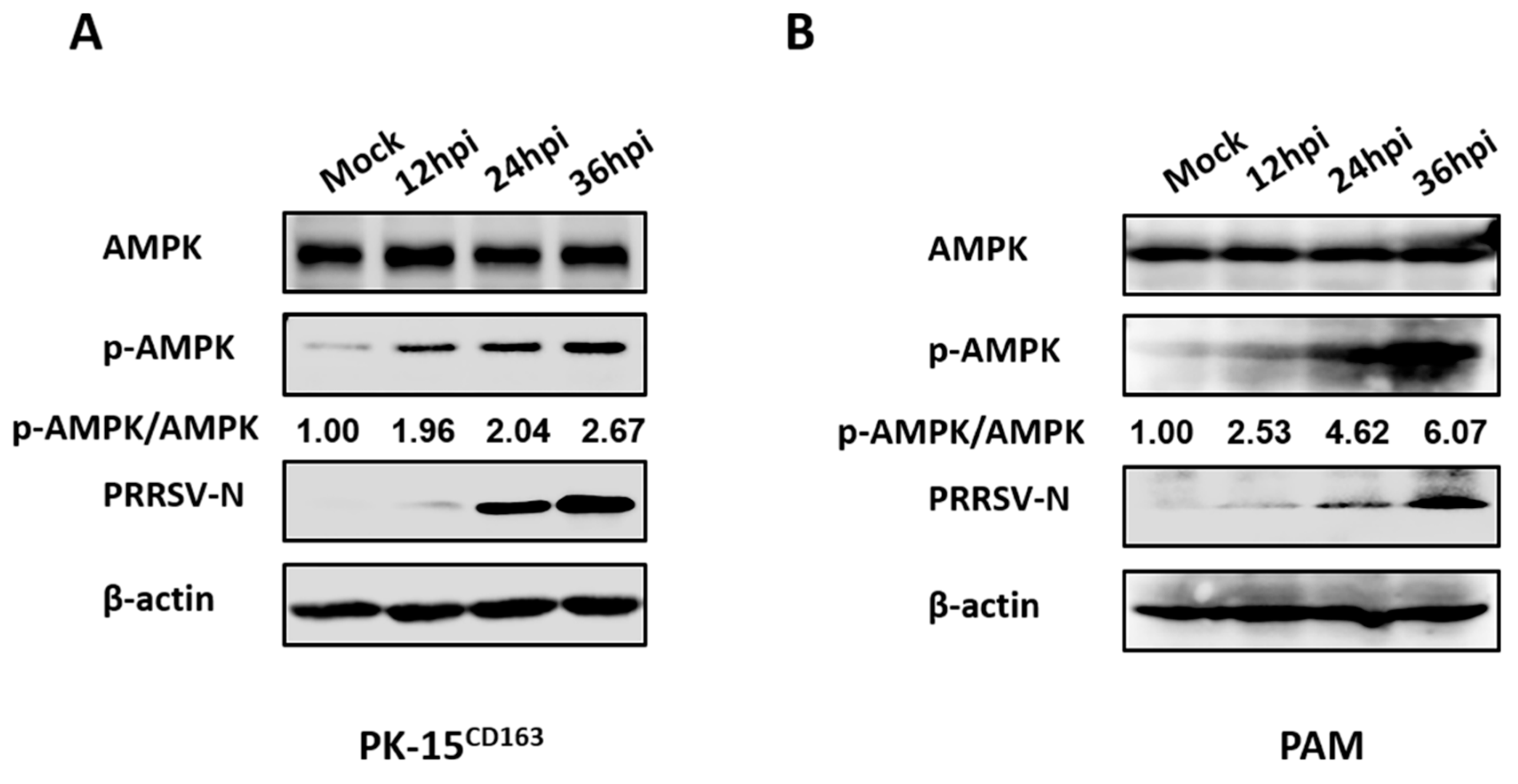
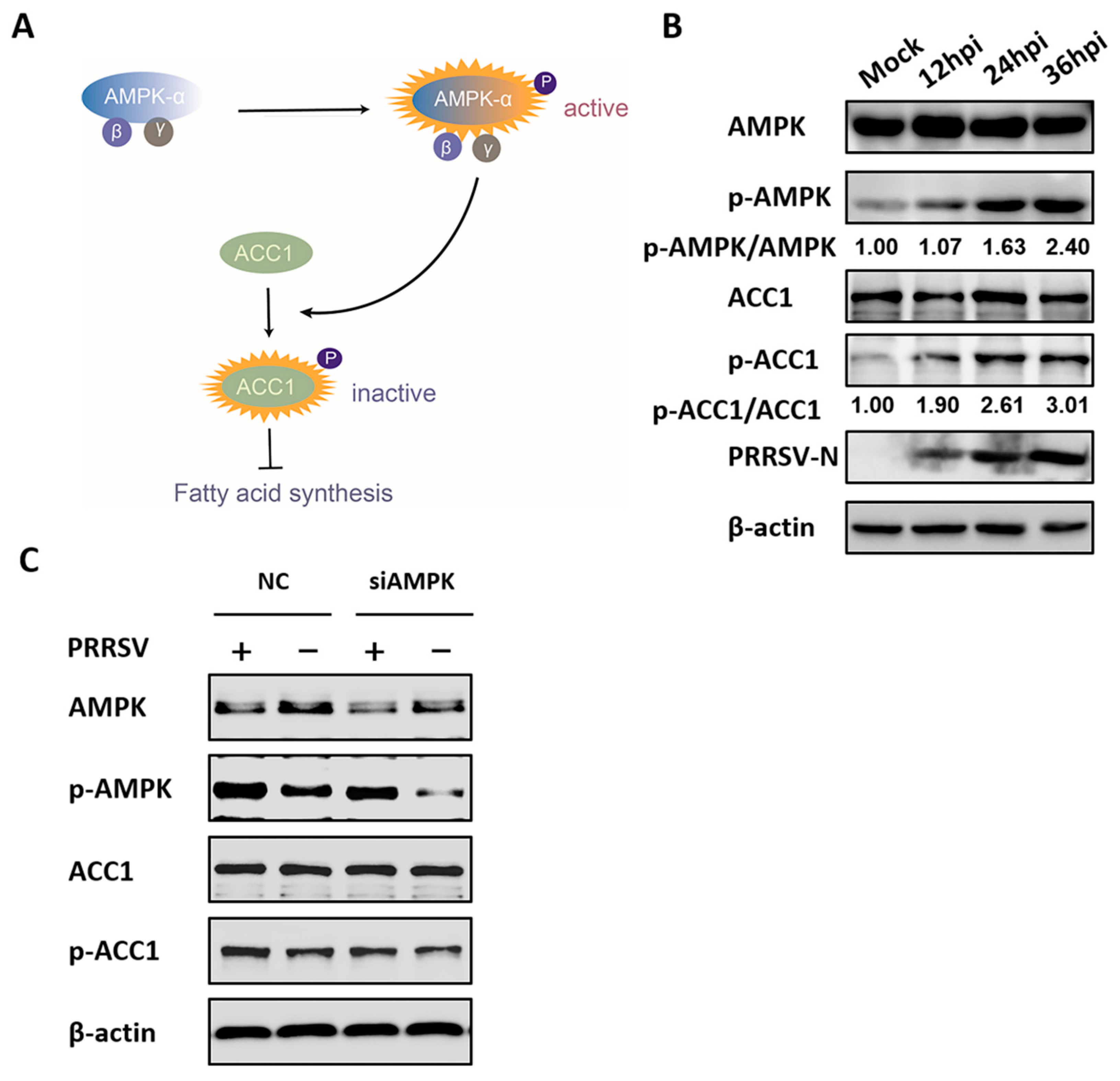
3.2. PRRSV Infection Upregulates AMPK Activity
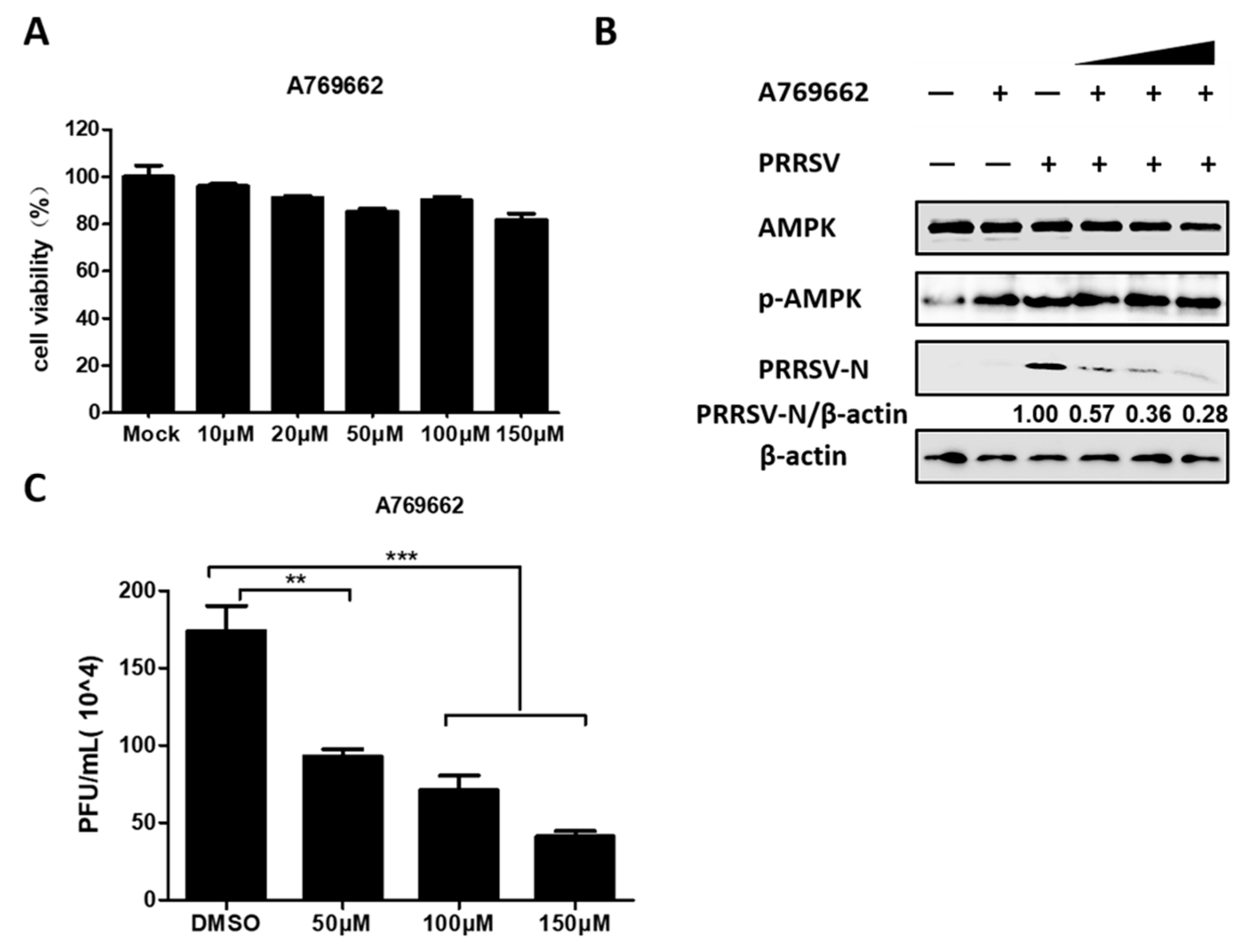
3.3. AMPK Activation Restricts PRRSV Infection
3.4. AMPK is Involved in PRRSV-Mediated ACC1 Activity Reduction
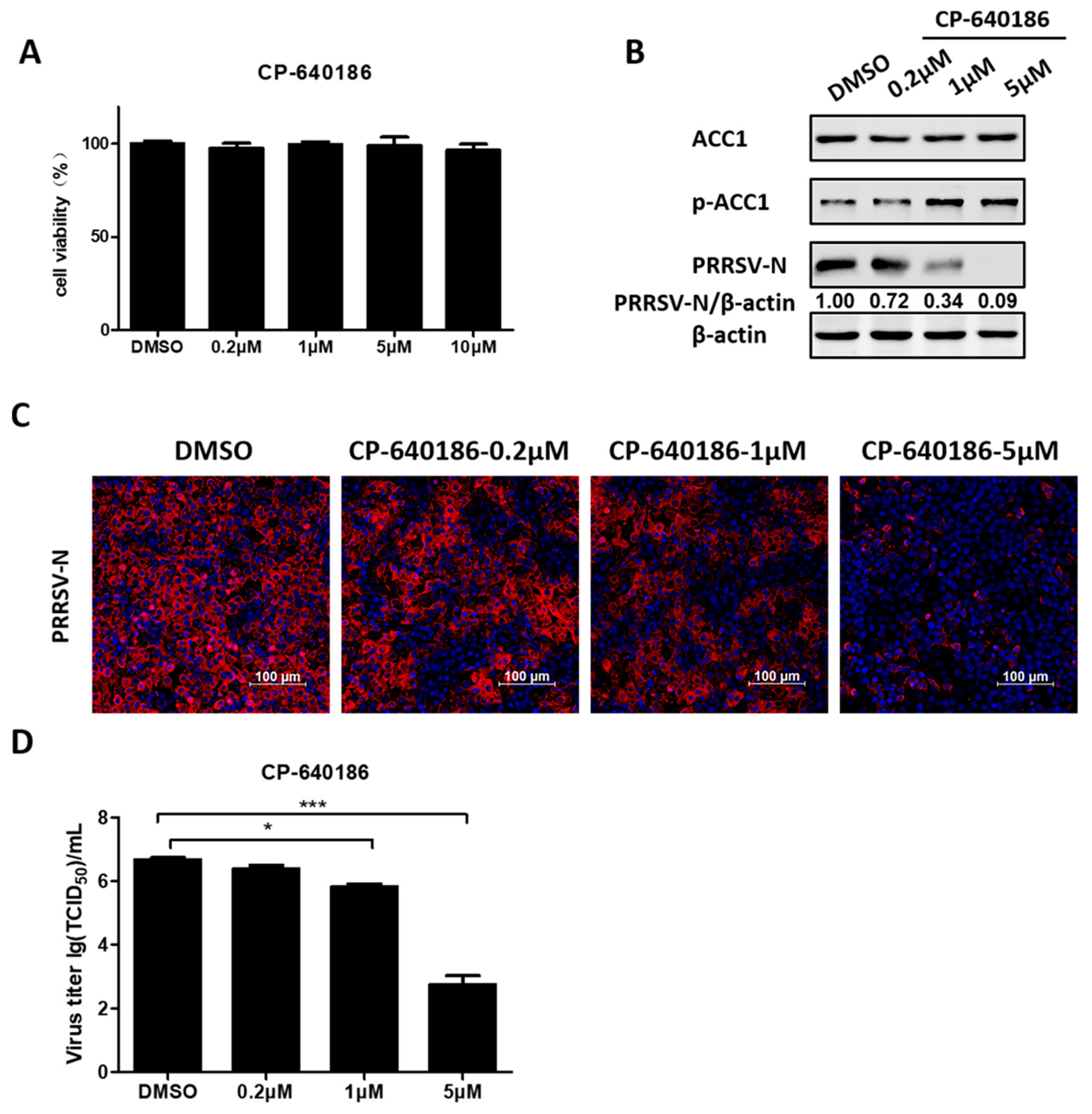
3.5. ACC1 Inhibitor Disrupts PRRSV Replication
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lunney, J.K.; Benfield, D.A.; Rowland, R.R.R. Porcine reproductive and respiratory syndrome virus: An update on an emerging and re-emerging viral disease of swine. Virus Res. 2010, 154, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2017, 209, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Kikkert, M.; Fang, Y. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 2013, 94, 2141–2163. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Gu, G.; Sun, W.; Zhang, Y.J.; Zhou, E.M. Improved vaccine against PRRSV: Current progress and future perspective. Front. Microbiol. 2017, 8, 1635. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shyu, D.L.; Shang, P.; Bai, J.; Ouyang, K.; Dhakal, S.; Hiremath, J.; Binjawadagi, B.; Renukaradhya, G.J.; Fang, Y. Mutations in a highly conserved motif of nsp1beta protein attenuate the innate immune suppression function of Porcine Reproductive and Respiratory Syndrome Virus. J. Virol. 2016, 90, 3584–3599. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.W.; Meng, X.J. Novel strategies and approaches to develop the next generation of vaccines against porcine reproductive and respiratory syndrome virus (PRRSV). Virus Res. 2010, 154, 141–149. [Google Scholar] [CrossRef]
- Rossow, K.D. Porcine Reproductive and Respiratory Syndrome. Vet. Pathol. 1998, 35, 1–20. [Google Scholar] [CrossRef]
- Mazzon, M.; Mercer, J. Lipid interactions during virus entry and infection. Cell. Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Fernandez de Castro, I.; Tenorio, R.; Risco, C. Virus assembly factories in a lipid world. Curr. Opin. Virol. 2016, 18, 20–26. [Google Scholar] [CrossRef]
- Dutta, A.; Sharma-Walia, N. Curbing lipids: Impacts on cancer and viral infection. Int. J. Mol. Sci. 2019, 20, 644. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Acosta, R.; Mosso, C.; Cervantes-Salazar, M.; Puerta-Guardo, H.; Medina, F.; Favari, L.; Ludert, J.E.; del Angel, R.M. The increase in cholesterol levels at early stages after dengue virus infection correlates with an augment in LDL particle uptake and HMG-CoA reductase activity. Virology 2013, 442, 132–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordier, B.B.; Ohkanda, J.; Liu, P.; Lee, S.Y.; Salazar, F.H.; Marion, P.L.; Ohashi, K.; Meuse, L.; Kay, M.A.; Casey, J.L.; et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J. Clin. Investig. 2003, 112, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Clark, P.J.; Thompson, A.J.; Vock, D.M.; Kratz, L.E.; Tolun, A.A.; Muir, A.J.; McHutchison, J.G.; Subramanian, M.; Millington, D.M.; Kelley, R.I.; et al. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology 2012, 56, 49–56. [Google Scholar] [CrossRef]
- Del Real, G.; Jimenez-Baranda, S.; Mira, E.; Lacalle, R.A.; Lucas, P.; Gomez-Mouton, C.; Alegret, M.; Pena, J.M.; Rodriguez-Zapata, M.; Alvarez-Mon, M.; et al. Statins inhibit HIV-1 infection by down-regulating Rho activity. J. Exp. Med. 2004, 200, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Moser, T.S.; Schieffer, D.; Cherry, S. AMP-activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog. 2012, 8, e1002661. [Google Scholar] [CrossRef] [Green Version]
- Okamura, H.; Nio, Y.; Akahori, Y.; Kim, S.; Watashi, K.; Wakita, T.; Hijikata, M. Fatty acid biosynthesis is involved in the production of hepatitis B virus particles. Biochem. Biophys. Res. Commun. 2016, 475, 87–92. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Carling, D.; Clarke, P.R.; Zammit, V.A.; Hardie, D.G. Purification and characterization of the AMP-activated protein kinase. Eur. J. Biochem. 1989, 186, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Chhipa, R.R. Evolving lessons on the complex role of AMPK in normal physiology and cancer. Trends Pharmacol. Sci. 2016, 37, 192–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, W.; Morantte, I.; Rodrigues, A.P.; Manning, G.; Montminy, M.; Shaw, R.J.; Dillin, A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 2011, 470, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, D.; Yamaguchi, A. A critical role of SNF1A/dAMPKalpha (Drosophila AMP-activated protein kinase alpha) in muscle on longevity and stress resistance in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2010, 394, 112–118. [Google Scholar] [CrossRef]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wei, R.; Li, Q.; Liu, H.; Huang, B.; Gao, J.; Mu, Y.; Wang, C.; Hsu, W.H.; Hiscox, J.A.; et al. PK-15 cells transfected with porcine CD163 by PiggyBac transposon system are susceptible to porcine reproductive and respiratory syndrome virus. J. Virol. Methods 2013, 193, 383–390. [Google Scholar] [CrossRef]
- Li, B.; Fang, L.; Liu, S.; Zhao, F.; Jiang, Y.; He, K.; Chen, H.; Xiao, S. The genomic diversity of Chinese porcine reproductive and respiratory syndrome virus isolates from 1996 to 2009. Vet. Microbiol. 2010, 146, 226–237. [Google Scholar] [CrossRef]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [Green Version]
- Taube, S.; Jiang, M.; Wobus, C.E. Glycosphingolipids as receptors for non-enveloped viruses. Viruses 2010, 2, 1011–1049. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Ábel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar] [PubMed]
- Chukkapalli, V.; Heaton, N.S.; Randall, G. Lipids at the interface of virus-host interactions. Curr. Opin. Microbiol. 2012, 15, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, Y.P.; Yu, Y.L.; Sun, M.X.; Li, C.; Chen, P.Y.; Mao, X. Role of lipid rafts in porcine reproductive and respiratory syndrome virus infection in MARC-145 cells. Biochem. Biophys. Res. Commun. 2011, 414, 545–550. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lee, C. Cellular cholesterol is required for porcine nidovirus infection. Arch. Virol. 2017, 162, 3753–3767. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, S.; Wang, D.; Luo, R.; Li, B.; Chen, H.; Fang, L. Cellular membrane cholesterol is required for porcine reproductive and respiratory syndrome virus entry and release in MARC-145 cells. Sci. China Life Sci. 2011, 54, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Zhang, Q.; Tang, J.; Feng, W.H. Lipid rafts both in cellular membrane and viral envelope are critical for PRRSV efficient infection. Virology 2015, 484, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Models Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Van Meer, G.; Voelker Dr Fau-Feigenson, G.W.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef] [Green Version]
- Greseth, M.D.; Traktman, P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for Vaccinia virus infection. PLoS Pathog. 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, D.L.; Cook, L.; Nagi, M.N.; Suneja, S.K. The fatty acid chain elongation system of mammalian endoplasmic reticulum. Prog. Lipid Res. 1992, 31, 1–51. [Google Scholar] [CrossRef]
- Ohol, Y.M.; Wang, Z.; Kemble, G.; Duke, G. Direct Inhibition of Cellular Fatty Acid Synthase Impairs Replication of Respiratory Syncytial Virus and Other Respiratory Viruses. PLoS ONE 2015, 10, e0144648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez de Oya, N.; Esler, W.P.; Huard, K.; El-Kattan, A.F.; Karamanlidis, G.; Blazquez, A.B.; Ramos-Ibeas, P.; Escribano-Romero, E.; Louloudes-Lazaro, A.; Casas, J.; et al. Targeting host metabolism by inhibition of acetyl-Coenzyme A carboxylase reduces flavivirus infection in mouse models. Emerg. Microbes Infect. 2019, 8, 624–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Koutsoudakis, G.; Romero-Brey, I.; Berger, C.; Pérez-Vilaró, G.; Monteiro Perin, P.; Vondran, F.W.R.; Kalesse, M.; Harmrolfs, K.; Müller, R.; Martinez, J.P.; et al. Soraphen A: A broad-spectrum antiviral natural product with potent anti-hepatitis C virus activity. J. Hepatol. 2015, 63, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Nio, Y.; Hasegawa, H.; Okamura, H.; Miyayama, Y.; Akahori, Y.; Hijikata, M. Liver-specific mono-unsaturated fatty acid synthase-1 inhibitor for anti-hepatitis C treatment. Antivir. Res. 2016, 132, 262–267. [Google Scholar] [CrossRef]
- Kuhn, R.J.; Soto-Acosta, R.; Bautista-Carbajal, P.; Cervantes-Salazar, M.; Angel-Ambrocio, A.H.; del Angel, R.M. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: A potential antiviral target. PLoS Pathog. 2017, 13, e1006257. [Google Scholar]
- Lo, A.K.F.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1–AMPK pathway by the Epstein–Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef]
- Mankouri, J.; Tedbury, P.R.; Gretton, S.; Hughes, M.E.; Griffin, S.D.C.; Dallas, M.L.; Green, K.A.; Hardie, D.G.; Peers, C.; Harris, M. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 11549–11554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.W.; Sun, L.J.; Liu, W.; Zhao, Y.H.; Kang, P.; Yan, B.Z. Hepatitis C virus core protein induces hepatic metabolism disorders through down-regulation of the SIRT1–AMPK signaling pathway. Int. J. Infect. Dis. 2013, 17, e539–e545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArdle, J.; Moorman, N.J.; Munger, J. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. 2012, 8, e1002502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdy, J.G.; Shenk, T.; Rabinowitz, J.D. Fatty acid elongase 7 catalyzes lipidome remodeling essential for human cytomegalovirus replication. Cell Rep. 2015, 10, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laufman, O.; Perrino, J.; Andino, R. Viral generated inter-organelle contacts redirect lipid flux for genome replication. Cell 2019, 178, 275–289. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293s–301s. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | siRNA Sequence (Sense) | siRNA Sequence (Anti-Sense) |
|---|---|---|
| Si-AMPK-1 | 5′-GGUUCUCAGCUGCCUUUAUTT-3′ | 5′-AUAAAGGCAGCUGAGAACCTT-3′ |
| Si-AMPK-2 | 5′-GCUUGCCAAAGGAAUGAUUTT-3′ | 5′-AAUCAUUCCUUUGGCAAGCTT-3′ |
| NC | 5′-UUCUCCGAACGUGUCACGUTT-3′ | 5′-ACGUGACACGUUCGGAGAATT-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, S.; Zhou, Y.; Bai, D.; Hao, W.; Zheng, B.; Xiao, S.; Fang, L. Fatty Acids Regulate Porcine Reproductive and Respiratory Syndrome Virus Infection via the AMPK-ACC1 Signaling Pathway. Viruses 2019, 11, 1145. https://0-doi-org.brum.beds.ac.uk/10.3390/v11121145
Long S, Zhou Y, Bai D, Hao W, Zheng B, Xiao S, Fang L. Fatty Acids Regulate Porcine Reproductive and Respiratory Syndrome Virus Infection via the AMPK-ACC1 Signaling Pathway. Viruses. 2019; 11(12):1145. https://0-doi-org.brum.beds.ac.uk/10.3390/v11121145
Chicago/Turabian StyleLong, Siwen, Yanrong Zhou, Dongcheng Bai, Wanjun Hao, Bohan Zheng, Shaobo Xiao, and Liurong Fang. 2019. "Fatty Acids Regulate Porcine Reproductive and Respiratory Syndrome Virus Infection via the AMPK-ACC1 Signaling Pathway" Viruses 11, no. 12: 1145. https://0-doi-org.brum.beds.ac.uk/10.3390/v11121145