Novel Viruses Found in Antricola Ticks Collected in Bat Caves in the Western Amazonia of Brazil


 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Ticks
2.2. Nucleic Acid Extraction
2.3. Nucleic Acid Amplification and Sequencing
2.4. High-Throughput Sequencing (HTS) Data Analysis
2.5. Viral Genetic Analyses
3. Results
3.1. Sequencing Output
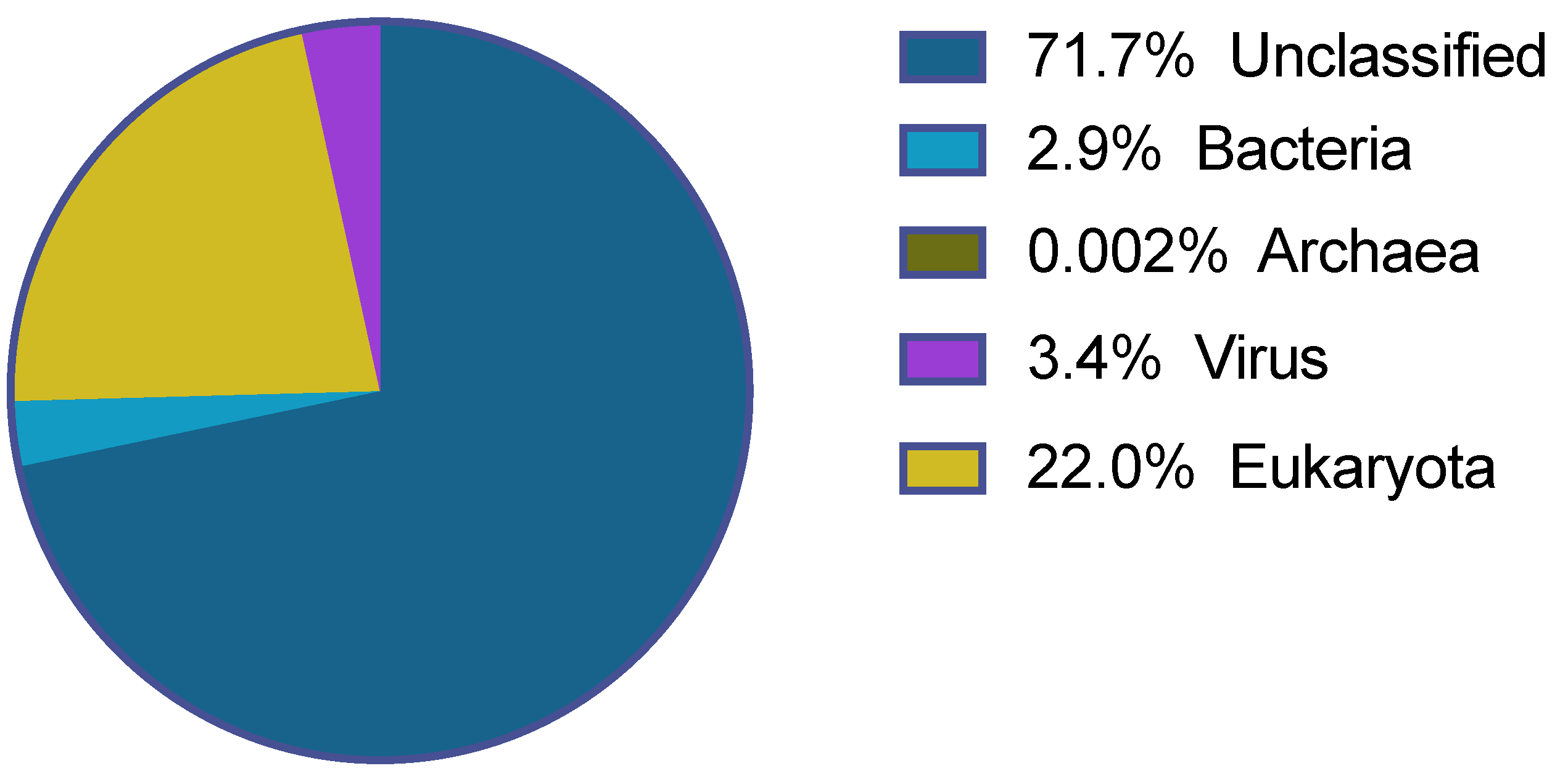
3.2. Viral Community
3.3. Single-Stranded Negative-Sense RNA Viruses
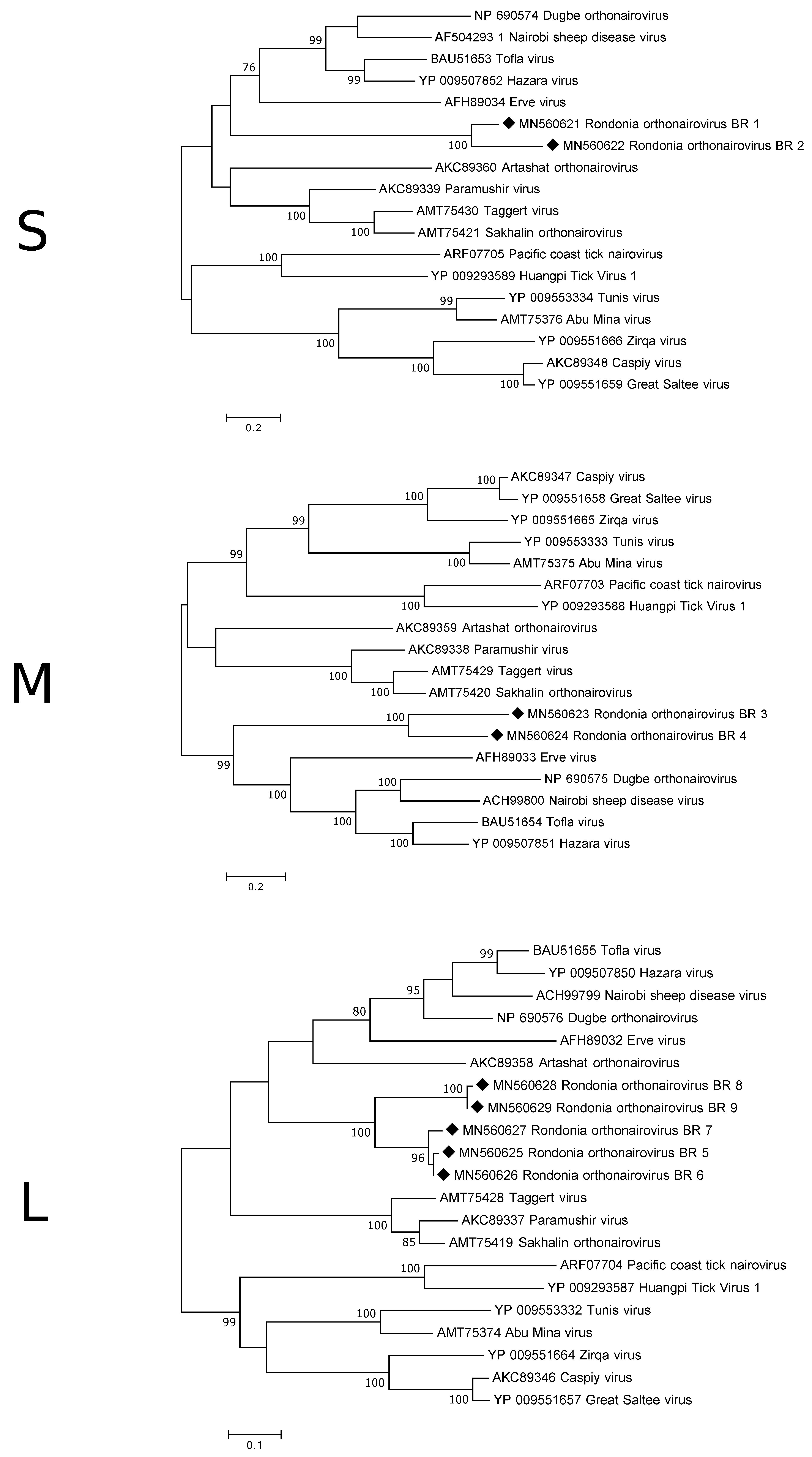
3.3.1. Nairoviridae
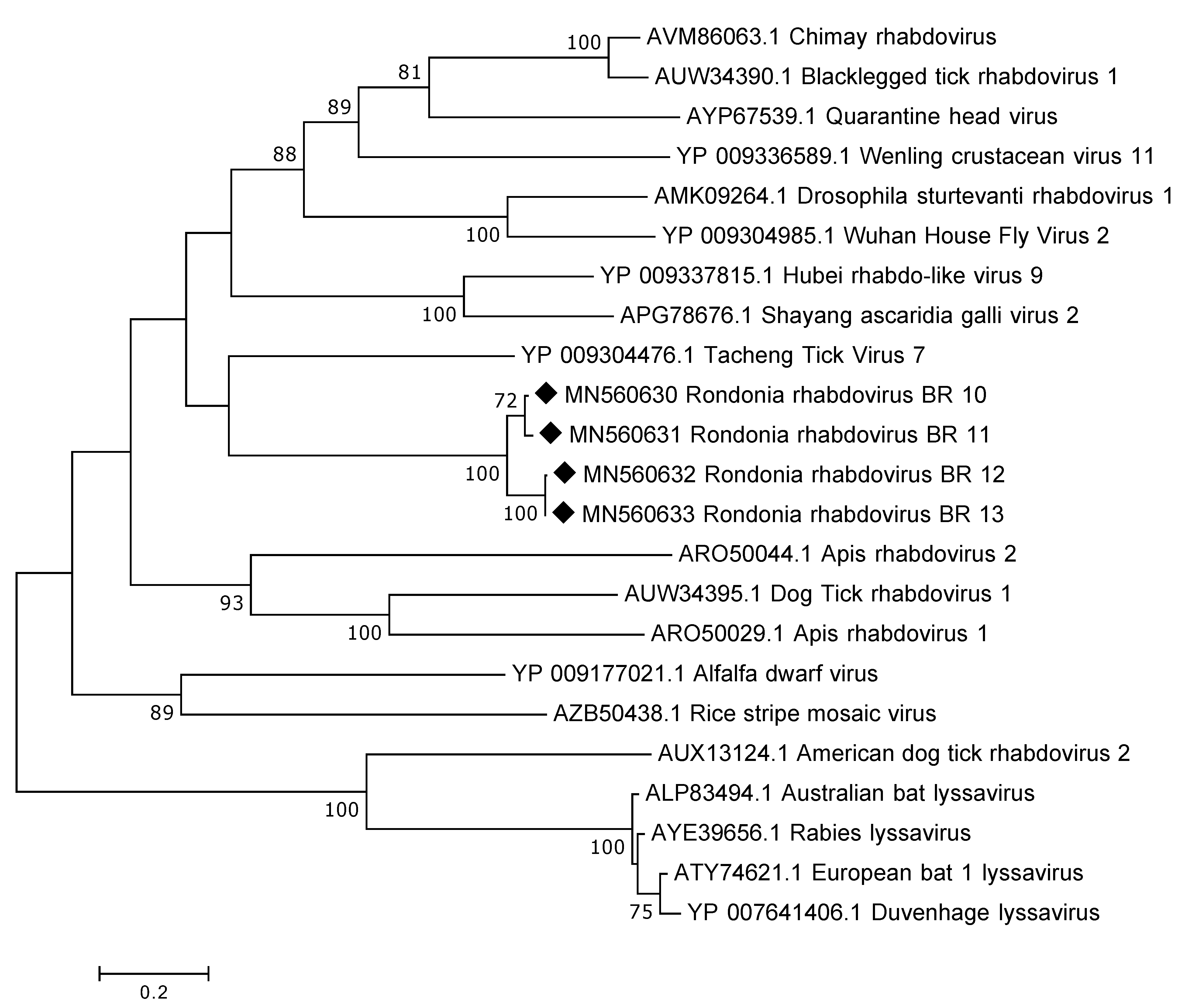
3.3.2. Rhabdoviridae
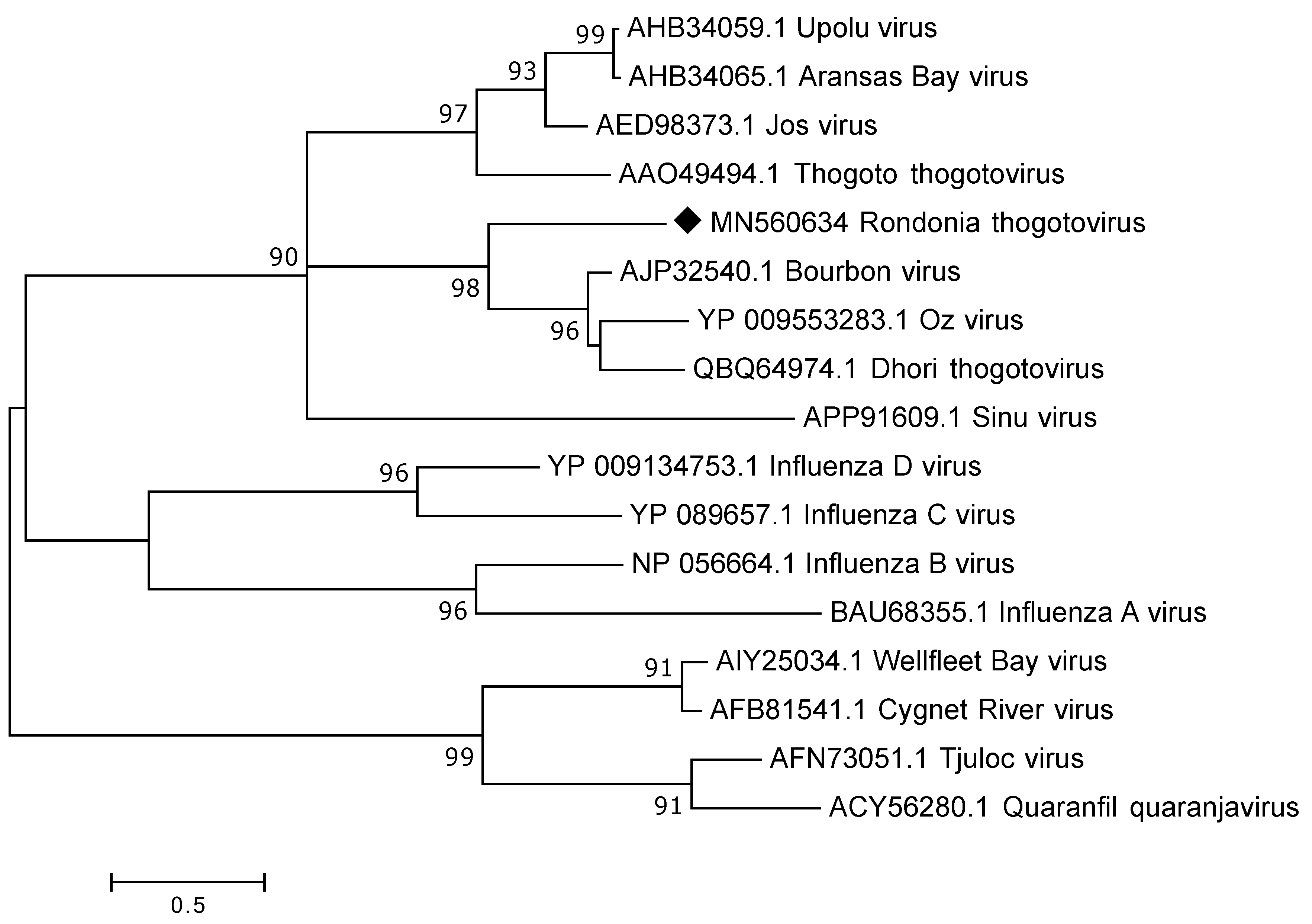
3.3.3. Orthomyxoviridae
3.4. Single-Stranded Positive-Sense RNA Viruses
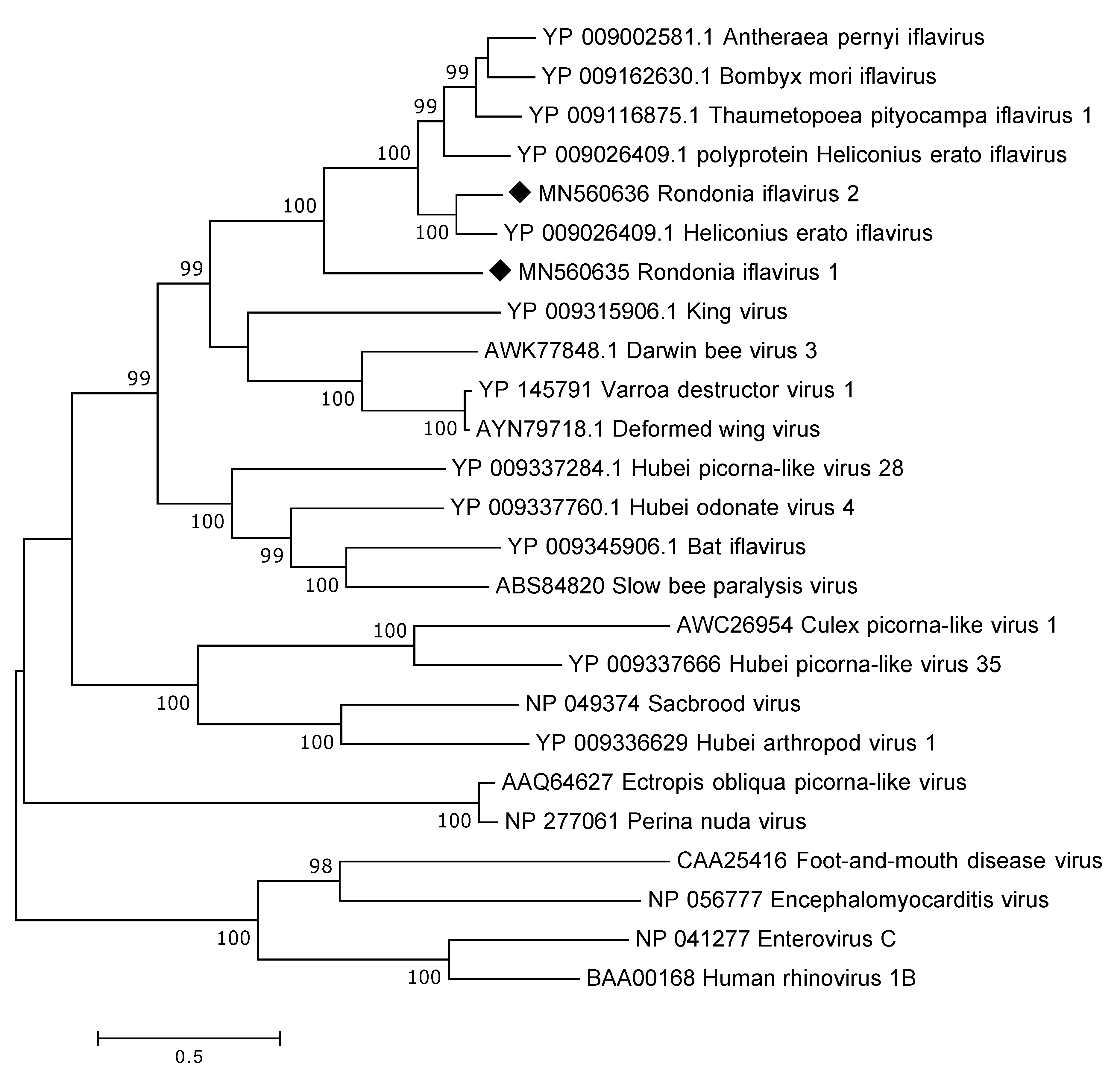
3.4.1. Iflaviridae
3.4.2. Dicistroviridae
3.4.3. Polycipiviridae
3.5. Double-Stranded RNA Viruses
Reoviridae
3.6. Unclassified Viruses
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weaver, S.C.; Reisen, W.K. Present and future arboviral threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef] [Green Version]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Mandl, J.N.; Schneider, C.; Schneider, D.S.; Baker, M.L. Going to Bat(s) for Studies of Disease Tolerance. Front. Immunol. 2018, 9, 2112. [Google Scholar] [CrossRef] [Green Version]
- Fagre, A.C.; Kading, R.C. Can Bats Serve as Reservoirs for Arboviruses? Viruses 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, W.M.; Fumagalli, M.J.; Torres Carrasco, A.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef] [PubMed]
- Cholleti, H.; Hayer, J.; Mulandane, F.C.; Falk, K.; Fafetine, J.; Berg, M.; Blomstrom, A.L. Viral metagenomics reveals the presence of highly divergent quaranjavirus in Rhipicephalus ticks from Mozambique. Infect. Ecol. Epidemiol. 2018, 8, 1478585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Hu, C.; Zhang, D.; Tang, S.; Zhang, Z.; Kou, Z.; Fan, Z.; Bente, D.; Zeng, C.; Li, T. Metagenomic profile of the viral communities in Rhipicephalus spp. ticks from Yunnan, China. PLoS ONE 2015, 10, e0121609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horak, I.G.; Camicas, J.L.; Keirans, J.E. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida): A world list of valid tick names. Exp. Appl. Acarol. 2002, 28, 27–54. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Sun, H.; Roberts, H. African swine fever. Antivir. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef]
- Hawman, D.W.; Feldmann, H. Recent advances in understanding Crimean-Congo hemorrhagic fever virus. F1000Research 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Valarcher, J.F.; Hagglund, S.; Juremalm, M.; Blomqvist, G.; Renstrom, L.; Zohari, S.; Leijon, M.; Chirico, J. Tick-borne encephalitis. Rev. Sci. Tech. 2015, 34, 453–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nava, S.; Venzal, J.M.; Terassini, F.A.; Mangold, A.J.; Camargo, L.M.; Labruna, M.B. Description of a new argasid tick (Acari: Ixodida) from bat caves in Brazilian Amazon. J. Parasitol. 2010, 96, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Guglielmone, A.A.; Venzal, J.M.; Battesti, D.; Onofrio, V.C.; Trajano, E.; Firminho, J.V.L. Ticks (Acari: Ixodida) of the Neotropical Zoogeographic Region; Consortium on Ticks and Tick-borne Diseases (ICTTD-2): Houten, The Netherlands, 2003; p. 173. [Google Scholar]
- Estrada-Pena, A.; Manuel Venzal, J.; Barros-Battesti, D.M.; Castilho Onofrio, V.; Trajano, E.; Lima Firmino, J.V. Three new species of Antricola (Acari: Argasidae) from Brazil, with a key to the known species in the genus. J. Parasitol. 2004, 90, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.M.; Labruna, M.B.; Mans, B.J.; Maruyama, S.R.; Francischetti, I.M.; Barizon, G.C.; de Miranda Santos, I.K. The sialotranscriptome of Antricola delacruzi female ticks is compatible with non-hematophagous behavior and an alternative source of food. Insect Biochem. Mol. Biol. 2012, 42, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, J.K.; Roark, A.M. Composition of guano produced by frugivorous, sanguivorous, and insectivorous bats. Acta Chiropterol. 2007, 9, 261–267. [Google Scholar] [CrossRef]
- Estrada-Barcenas, D.A.; Palacios-Vargas, J.G.; Estrada-Venegas, E.; Klimov, P.B.; Martinez-Mena, A.; Taylor, M.L. Biological activity of the mite Sancassania sp. (Acari: Acaridae) from bat guano associated with the pathogenic fungus Histoplasma capsulatum. Mem. Inst. Oswaldo Cruz 2010, 105, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Labruna, M.B.; Nava, S.; Terassini, F.A.; Onofrio, V.C.; Barros-Battesti, D.M.; Camargo, L.M.; Venzal, J.M. Description of adults and nymph, and redescription of the larva, of Ornithodoros marinkellei (Acari:Argasidae), with data on its phylogenetic position. J. Parasitol. 2011, 97, 207–217. [Google Scholar] [CrossRef]
- Labruna, M.B.; Terassini, F.A.; Camargo, L.M.; Brandao, P.E.; Ribeiro, A.F.; Estrada-Pena, A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J. Parasitol. 2008, 94, 788–792. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Gorska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Vandegrift, K.J.; Kapoor, A. The Ecology of New Constituents of the Tick Virome and Their Relevance to Public Health. Viruses 2019, 11, 529. [Google Scholar] [CrossRef] [Green Version]
- Lasecka, L.; Baron, M.D. The molecular biology of nairoviruses, an emerging group of tick-borne arboviruses. Arch. Virol. 2014, 159, 1249–1265. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Banyard, A.C.; Evans, J.S.; Luo, T.R.; Fooks, A.R. Lyssaviruses and bats: Emergence and zoonotic threat. Viruses 2014, 6, 2974–2990. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4. [Google Scholar] [CrossRef]
- Haig, D.A.; Woodall, J.P.; Danskin, D. Thogoto Virus—A Hitherto Undescribed Agent Isolated from Ticks in Kenya. J. Gen. Microbiol. 1965, 38, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Primers 2017, 3, 17083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desselberger, U. Rotaviruses. Virus Res. 2014, 190, 75–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Type | Viral Family | Number of Assigned Reads | % of Viral Reads |
|---|---|---|---|
| Single-stranded negative-sense RNA virus | Nairoviridae | 753,197 | 88.2 |
| Peribunyaviridae | 10,515 | 1.2 | |
| Rhabdoviridae | 11,075 | 1.3 | |
| Orthomyxoviridae | 17,553 | 2.1 | |
| Single-stranded positive-sense RNA virus | Iflaviridae | 14,040 | 1.6 |
| Dicistroviridae | 10,224 | 1.2 | |
| Polycipiviridae | 1271 | 0.1 | |
| Double-stranded RNA virus | Reoviridae | 702 | 0.1 |
| Unclassified viruses | Unclassified viruses | 32,371 | 3.8 |
| Segment | Protein | % Coverage | % a.a. Identity |
|---|---|---|---|
| 1 | PB2 | 68 | 97 |
| 2 | PB1 | 92 | 82 |
| 3 | PA | 47 | 54 |
| 4 | NP | 73 | 61 |
| 5 | GP | 38 | 53 |
| 6 | M | 100 | 47 |
| Segment | Protein | % Cover | % a.a. Identity |
|---|---|---|---|
| 1 | VP1 | 88.5 | 74–87 |
| 2 | VP2 | 75.7 | 60–94 |
| 3 | VP3 | 38.9 | 55–59 |
| 4 | VP4 | 62.7 | 64 |
| 5 | NSP1 | 67.3 | 52–57 |
| 6 | VP6 | 67.9 | 84 |
| 7 | NSP3 | 76.0 | 63 |
| 8 | NSP2 | 92.6 | 79–81 |
| 9 | VP7 | 54.7 | 63 |
| 10 | NSP4 | 27.7 | 49–53 |
| 11 | NSP5 | 87.5 | 61 |
| 11 | NSP6 | n.d. | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blomström, A.-L.; Luz, H.R.; Öhlund, P.; Lukenge, M.; Brandão, P.E.; Labruna, M.B.; Berg, M. Novel Viruses Found in Antricola Ticks Collected in Bat Caves in the Western Amazonia of Brazil. Viruses 2020, 12, 48. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010048
Blomström A-L, Luz HR, Öhlund P, Lukenge M, Brandão PE, Labruna MB, Berg M. Novel Viruses Found in Antricola Ticks Collected in Bat Caves in the Western Amazonia of Brazil. Viruses. 2020; 12(1):48. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010048
Chicago/Turabian StyleBlomström, Anne-Lie, Hermes R. Luz, Pontus Öhlund, Matthew Lukenge, Paulo Eduardo Brandão, Marcelo B. Labruna, and Mikael Berg. 2020. "Novel Viruses Found in Antricola Ticks Collected in Bat Caves in the Western Amazonia of Brazil" Viruses 12, no. 1: 48. https://0-doi-org.brum.beds.ac.uk/10.3390/v12010048