Genetic and Biological Diversity of Porcine Sapeloviruses Prevailing in Zambia



 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Detection of PSV by RT-PCR
2.3. Cells and Virus Isolation
2.4. Whole Genome Sequencing
2.5. Phylogenetic Analysis and Other Bioinformatic Analyses
2.6. Growth Kinetics of PSV
2.7. Statistical Analysis
3. Results
3.1. Prevalence of PSV Infection in Zambia
3.2. Isolation and Full Genome Sequence of PSVs
3.3. Comparative Genome Analysis among PSVs
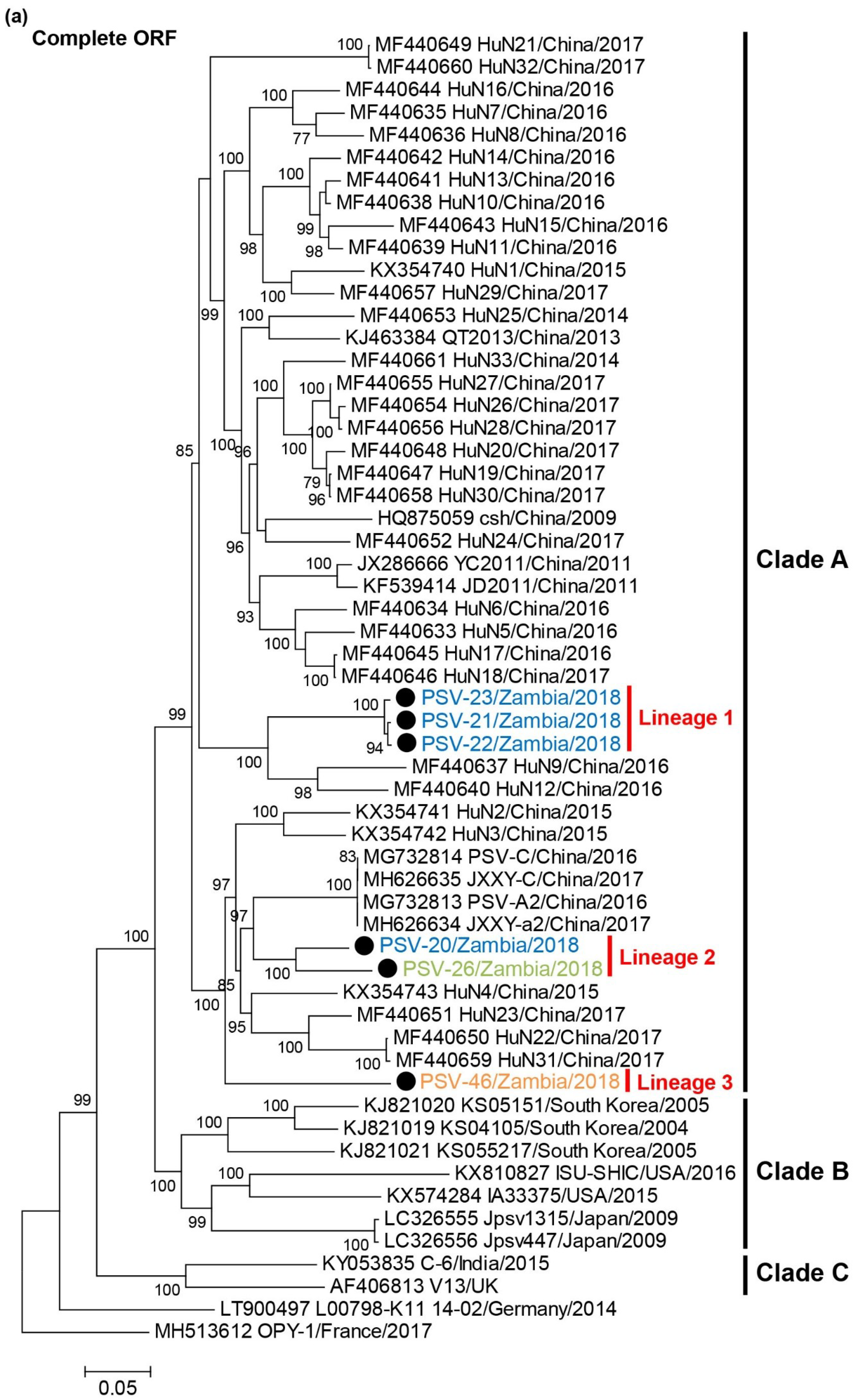
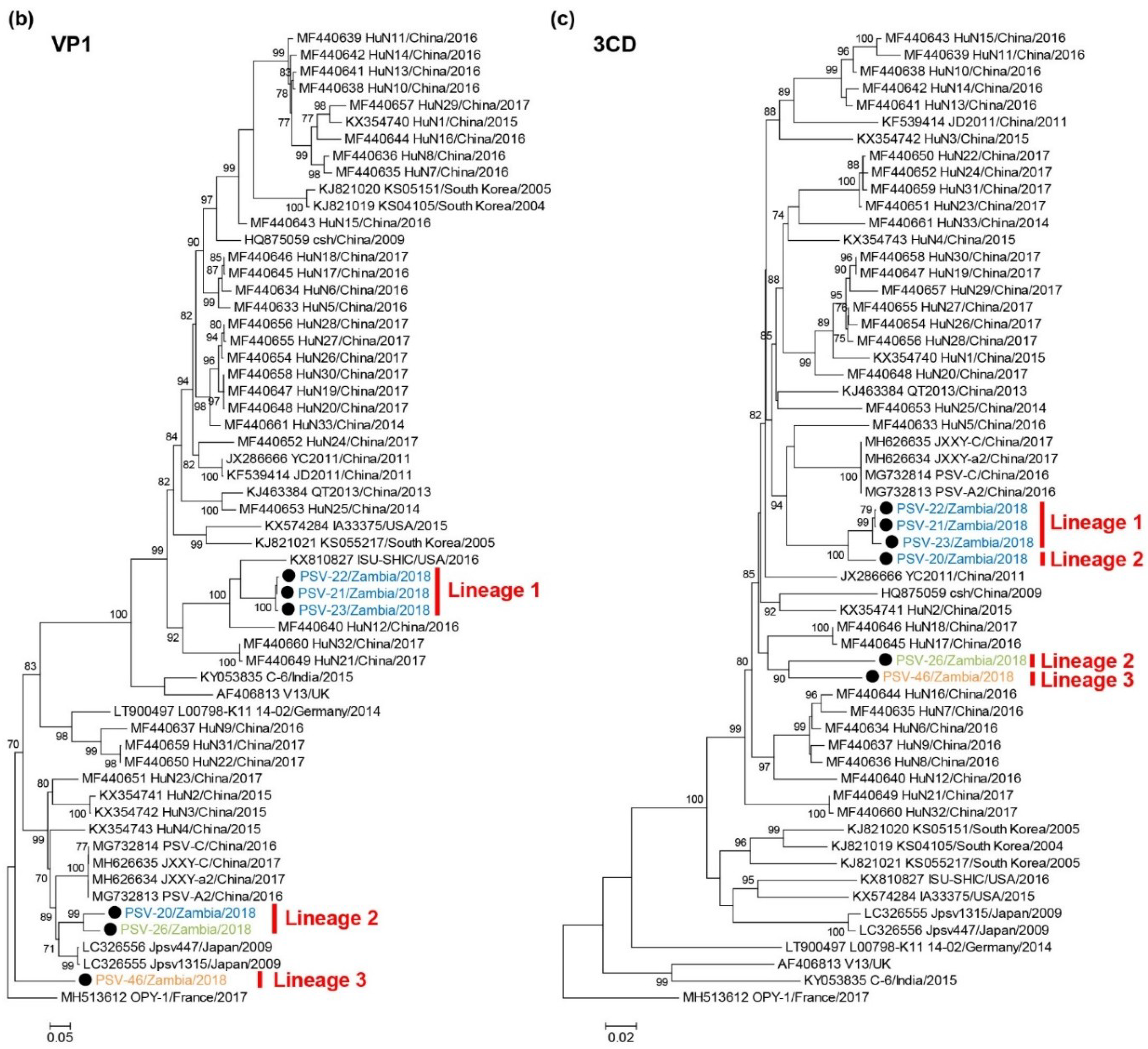
3.4. Phylogenetic Analyses of PSVs
3.5. Recombination Analyses of PSVs
3.6. Growth Kinetics of Zambian PSVs In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Dunne, H.W.; Wang, J.T.; Ammerman, E.H. Classification of North American porcine enteroviruses: A comparison with European and Japanese strains. Infect. Immun. 1971, 4, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racaniello, V.R. Picornaviridae: The Viruses and Their Replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Wolters Kluwer/Lippincott Williams, & Winlkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 453–489. [Google Scholar]
- Arruda, P.H.; Arruda, B.L.; Schwartz, K.J.; Vannucci, F.; Resende, T.; Rovira, A.; Sundberg, P.; Nietfeld, J.; Hause, B.M. Detection of a novel sapelovirus in central nervous tissue of pigs with polioencephalomyelitis in the USA. Transbound. Emerg. Dis. 2017, 64, 311–315. [Google Scholar] [CrossRef] [PubMed]
- LAMONT, P.H.; BETTS, A.O. Enteroviruses in the pig. Nature 1958, 182, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Ji, W.; Yang, S.; Cui, L.; Yang, Z.; Yuan, C.; Hua, X. Isolation and characterization of the first Chinese porcine sapelovirus strain. Arch. Virol. 2011, 156, 1567–1574. [Google Scholar] [CrossRef]
- Li, Y.; Du, L.; Jin, T.; Cheng, Y.; Zhang, X.; Jiao, S.; Huang, T.; Zhang, Y.; Yan, Y.; Gu, J.; et al. Characterization and epidemiological survey of porcine sapelovirus in China. Vet. Microbiol. 2019, 232, 13–21. [Google Scholar] [CrossRef]
- Ray, P.K.; Desingu, P.A.; Kumari, S.; John, J.K.; Sethi, M.; Sharma, G.K.; Pattnaik, B.; Singh, R.K.; Saikumar, G. Porcine sapelovirus among diarrhoeic piglets in India. Transbound. Emerg. Dis. 2018, 65, 261–263. [Google Scholar] [CrossRef] [Green Version]
- Piorkowski, G.; Capai, L.; Falchi, A.; Casabianca, F.; Maestrini, O.; Gallian, P.; Barthélémy, K.; Py, O.; Charrel, R.; de Lamballerie, X. First Identification and Genomic Characterization of a Porcine Sapelovirus from Corsica, France, 2017. Microbiol. Resour. Announc. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Li, R.; Peng, W.; Ge, M.; Luo, B.; Qu, T.; Yu, X. First isolation and genetic characteristics of porcine sapeloviruses in Hunan, China. Arch. Virol. 2017, 162, 1589–1597. [Google Scholar] [CrossRef]
- Son, K.Y.; Kim, D.S.; Kwon, J.; Choi, J.S.; Kang, M.I.; Belsham, G.J.; Cho, K.O. Full-length genomic analysis of Korean porcine sapelovirus strains. PLoS ONE 2014, 9, e107860. [Google Scholar] [CrossRef]
- Hanke, D.; Pohlmann, A.; Sauter-Louis, C.; Höper, D.; Stadler, J.; Ritzmann, M.; Steinrigl, A.; Schwarz, B.A.; Akimkin, V.; Fux, R.; et al. Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology. Viruses 2017, 9, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Wang, L.; Zheng, Y.; Zhang, J.; Guo, B.; Yoon, K.J.; Gauger, P.C.; Harmon, K.M.; Main, R.G.; Li, G. Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virol. J. 2018, 15, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitrago, D.; Cano-Gómez, C.; Agüero, M.; Fernandez-Pacheco, P.; Gómez-Tejedor, C.; Jiménez-Clavero, M.A. A survey of porcine picornaviruses and adenoviruses in fecal samples in Spain. J. Vet. Diagn. Investig. 2010, 22, 763–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Liu, J.; Fang, L.; Kataoka, M.; Takeda, N.; Wakita, T.; Li, T.C. Characterization of porcine sapelovirus isolated from Japanese swine with PLC/PRF/5 cells. Transbound. Emerg. Dis. 2018, 65, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Donin, D.G.; de Arruda Leme, R.; Alfieri, A.F.; Alberton, G.C.; Alfieri, A.A. First report of Porcine teschovirus (PTV), Porcine sapelovirus (PSV) and Enterovirus G (EV-G) in pig herds of Brazil. Trop. Anim. Health. Prod. 2014, 46, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Prodělalová, J. The survey of porcine teschoviruses, sapeloviruses and enteroviruses B infecting domestic pigs and wild boars in the Czech Republic between 2005 and 2011. Infect. Genet. Evol. 2012, 12, 1447–1451. [Google Scholar] [CrossRef]
- Schock, A.; Gurrala, R.; Fuller, H.; Foyle, L.; Dauber, M.; Martelli, F.; Scholes, S.; Roberts, L.; Steinbach, F.; Dastjerdi, A. Investigation into an outbreak of encephalomyelitis caused by a neuroinvasive porcine sapelovirus in the United Kingdom. Vet. Microbiol. 2014, 172, 381–389. [Google Scholar] [CrossRef]
- Sunaga, F.; Masuda, T.; Ito, M.; Akagami, M.; Naoi, Y.; Sano, K.; Katayama, Y.; Omatsu, T.; Oba, M.; Sakaguchi, S.; et al. Complete genomic analysis and molecular characterization of Japanese porcine sapeloviruses. Virus Genes 2019, 55, 198–208. [Google Scholar] [CrossRef]
- Yang, T.; Yu, X.; Yan, M.; Luo, B.; Li, R.; Qu, T.; Luo, Z.; Ge, M.; Zhao, D. Molecular characterization of Porcine sapelovirus in Hunan, China. J. Gen. Virol. 2017, 98, 2738–2747. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, C.; Yue, H.; Ren, Y.; Song, Z. Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. J. Gen. Virol. 2014, 95, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Forman, A.J.; Pass, D.A.; Connaughton, I.D. The characterisation and pathogenicity of porcine enteroviruses isolated in Victoria. Aust. Vet. J. 1982, 58, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Tassoni, L.; Zamperin, G.; Monne, I.; Beato, M.S. Nearly Complete Genome Sequence of a Sapelovirus A Strain Identified in Swine in Italy. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amimo, J.O.; El Zowalaty, M.E.; Githae, D.; Wamalwa, M.; Djikeng, A.; Nasrallah, G.K. Metagenomic analysis demonstrates the diversity of the fecal virome in asymptomatic pigs in East Africa. Arch. Virol. 2016, 161, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kang, M.I.; Son, K.Y.; Bak, G.Y.; Park, J.G.; Hosmillo, M.; Seo, J.Y.; Kim, J.Y.; Alfajaro, M.M.; Soliman, M.; et al. Pathogenesis of Korean SapelovirusA in piglets and chicks. J. Gen. Virol. 2016, 97, 2566–2574. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Kitikoon, P.; Nilubol, D.; Erickson, B.J.; Janke, B.H.; Hoover, T.C.; Sornsen, S.A.; Thacker, E.L. The immune response and maternal antibody interference to a heterologous H1N1 swine influenza virus infection following vaccination. Vet. Immunol. Immunopathol. 2006, 112, 117–128. [Google Scholar] [CrossRef]
- Wang, W.H.; Lin, C.Y.; Chang Ishcol, M.R.; Urbina, A.N.; Assavalapsakul, W.; Thitithanyanont, A.; Lu, P.L.; Chen, Y.H.; Wang, S.F. Detection of African swine fever virus in pork products brought to Taiwan by travellers. Emerg. Microbes. Infect. 2019, 8, 1000–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Harada, R.; Okamatsu, M.; Matsuno, K.; Arita, T.; Suzuki, Y.; Shirakura, M.; Odagiri, T.; Takemae, N.; Uchida, Y.; et al. Characterization of a novel reassortant H7N3 highly pathogenic avian influenza virus isolated from a poultry meat product taken on a passenger flight to Japan. J. Vet. Med. Sci. 2019, 81, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Mase, M.; Eto, M.; Tanimura, N.; Imai, K.; Tsukamoto, K.; Horimoto, T.; Kawaoka, Y.; Yamaguchi, S. Isolation of a genotypically unique H5N1 influenza virus from duck meat imported into Japan from China. Virology 2005, 339, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmock, N.J.; Easton, A.J.; Leppard, K.N. Introduction to Modern Virology, 7th ed.; John Wiley & Sons: Chichester, UK, 2016; pp. 39–51. [Google Scholar]
- Ansardi, D.C.; Luo, M.; Morrow, C.D. Mutations in the poliovirus P1 capsid precursor at arginine residues VP4-ARG34, VP3-ARG223, and VP1-ARG129 affect virus assembly and encapsidation of genomic RNA. Virology 1994, 199, 20–34. [Google Scholar] [CrossRef]
- Danthi, P.; Tosteson, M.; Li, Q.H.; Chow, M. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J. Virol. 2003, 77, 5266–5274. [Google Scholar] [CrossRef] [Green Version]
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 43–89. [Google Scholar] [CrossRef]
- Ouyang, T.; Zhang, X.; Liu, X.; Ren, L. Co-Infection of Swine with Porcine Circovirus Type 2 and Other Swine Viruses. Viruses 2019, 11, 185. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm | District | Date | No. of Fecal Samples |
|---|---|---|---|
| A | Lusaka | 7 January, 14 June | 39 |
| B | Chilanga | 25 January, 10 July, 4 December | 30 |
| C | Kafue | 13 February, 8 June, 16 August | 58 |
| D | Chibombo | 2 March | 4 |
| E | Lusaka | 17 July, 20 December | 16 |
| Age (Week) | Stage | No. of Positive/No. of Tested Samples (%) | ||
|---|---|---|---|---|
| Healthy | Diarrheal | Total | ||
| 0–3 | Suckling | 3/7 (42.9) | 14/40 (35.0) | 17/47 (36.2) |
| 4–12 | Fattening | 39/41 (95.1) | 55/59 (93.2) | 94/100 (94.0) |
| Strain | Farm | District | Sampling Date | Cell Line 1 | Obtained Genome Length (nt) 2 | Length of Complete ORF (nt) |
|---|---|---|---|---|---|---|
| PSV-20-V | A | Lusaka | 7 January | Vero E6 | 7495 | 6999 |
| PSV-21-V | A | Lusaka | 7 January | Vero E6 | 7458 | 6972 |
| PSV-21-B | A | Lusaka | 7 January | BHK | 7468 | 6972 |
| PSV-22-B | A | Lusaka | 7 January | BHK | 7468 | 6972 |
| PSV-23-V | A | Lusaka | 7 January | Vero E6 | 7462 | 6972 |
| PSV-23-B | A | Lusaka | 7 January | BHK | 7462 | 6972 |
| PSV-26-B | B | Chilanga | 25 January | BHK | 7493 | 6996 |
| PSV-46-V | D | Chibombo | 2 March | Vero E6 | 7469 | 6972 |
| PSV-46-B | D | Chibombo | 2 March | BHK | 7498 | 6972 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harima, H.; Kajihara, M.; Simulundu, E.; Bwalya, E.; Qiu, Y.; Isono, M.; Okuya, K.; Gonzalez, G.; Yamagishi, J.; Hang’ombe, B.M.; et al. Genetic and Biological Diversity of Porcine Sapeloviruses Prevailing in Zambia. Viruses 2020, 12, 180. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020180
Harima H, Kajihara M, Simulundu E, Bwalya E, Qiu Y, Isono M, Okuya K, Gonzalez G, Yamagishi J, Hang’ombe BM, et al. Genetic and Biological Diversity of Porcine Sapeloviruses Prevailing in Zambia. Viruses. 2020; 12(2):180. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020180
Chicago/Turabian StyleHarima, Hayato, Masahiro Kajihara, Edgar Simulundu, Eugene Bwalya, Yongjin Qiu, Mao Isono, Kosuke Okuya, Gabriel Gonzalez, Junya Yamagishi, Bernard M. Hang’ombe, and et al. 2020. "Genetic and Biological Diversity of Porcine Sapeloviruses Prevailing in Zambia" Viruses 12, no. 2: 180. https://0-doi-org.brum.beds.ac.uk/10.3390/v12020180