Pervasive Differential Splicing in Marek’s Disease Virus Can Discriminate CVI-988 Vaccine Strain from RB-1B Very Virulent Strain in Chicken Embryonic Fibroblasts

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-Sequencing
2.1.1. Cell and Virus History
2.1.2. Sample Preparation
2.1.3. Sequencing
2.2. Bioinformatics Selection of Biomarkers
2.2.1. Primary Analysis
2.2.2. Tentative Annotation of Full Spliced Coding Transcripts
2.2.3. Selection of Relevant MDV Encoded Introns
2.2.4. Differential Expression of Transcripts
2.2.5. Data Visualisation
2.3. Real-Time PCR Validation of Viral Junctions
2.3.1. Sample Preparation
2.3.2. Primers and Probes
2.3.3. Relative Quantitative RT-PCR
2.3.4. Accession Numbers
3. Results
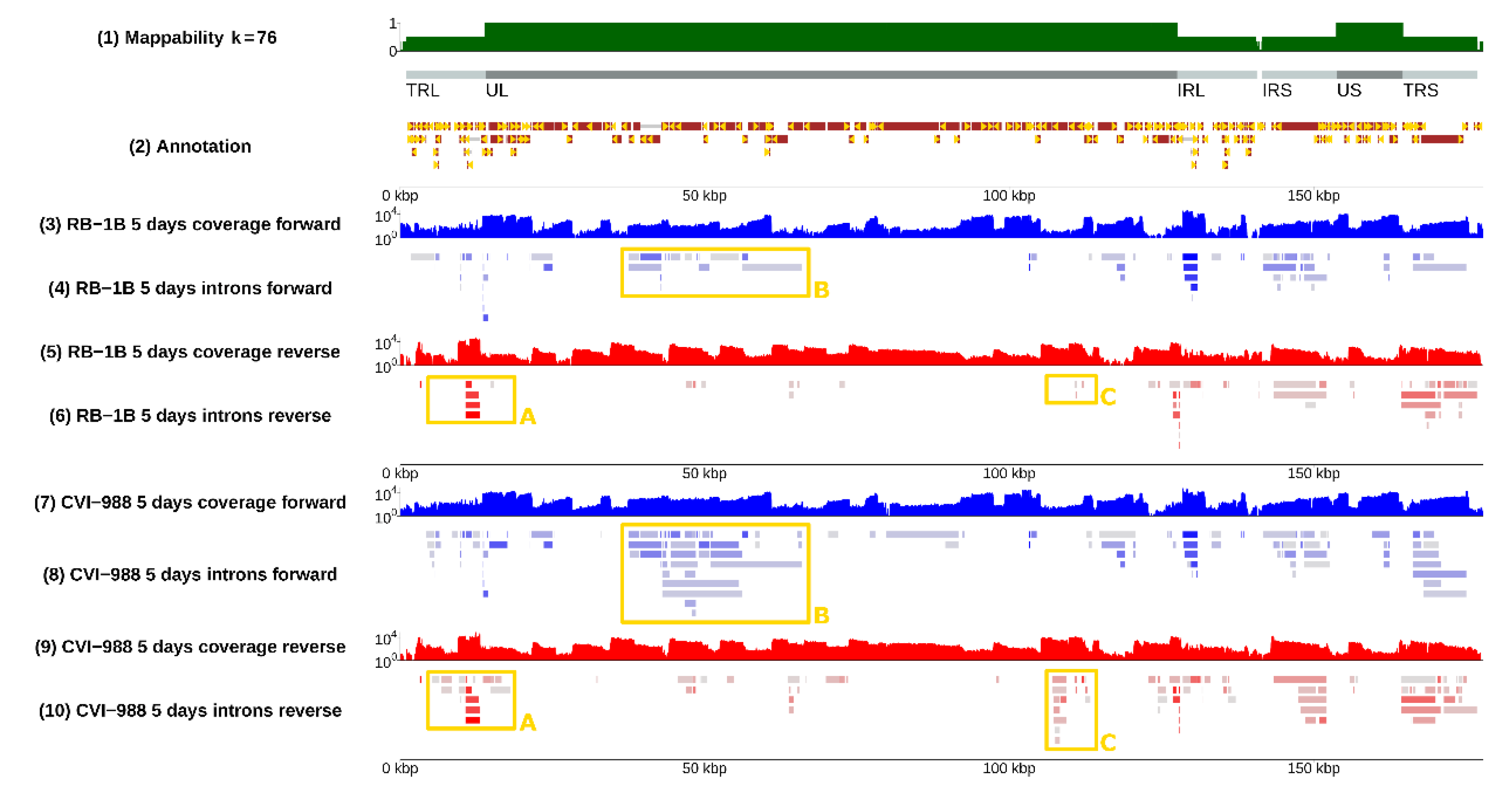
3.1. Splicing Was Pervasive in MDV
3.2. Splicing Was Strain-Specific, and Occurred More Frequently in CVI-988
3.3. Tentative Reannotation of Coding Transcripts
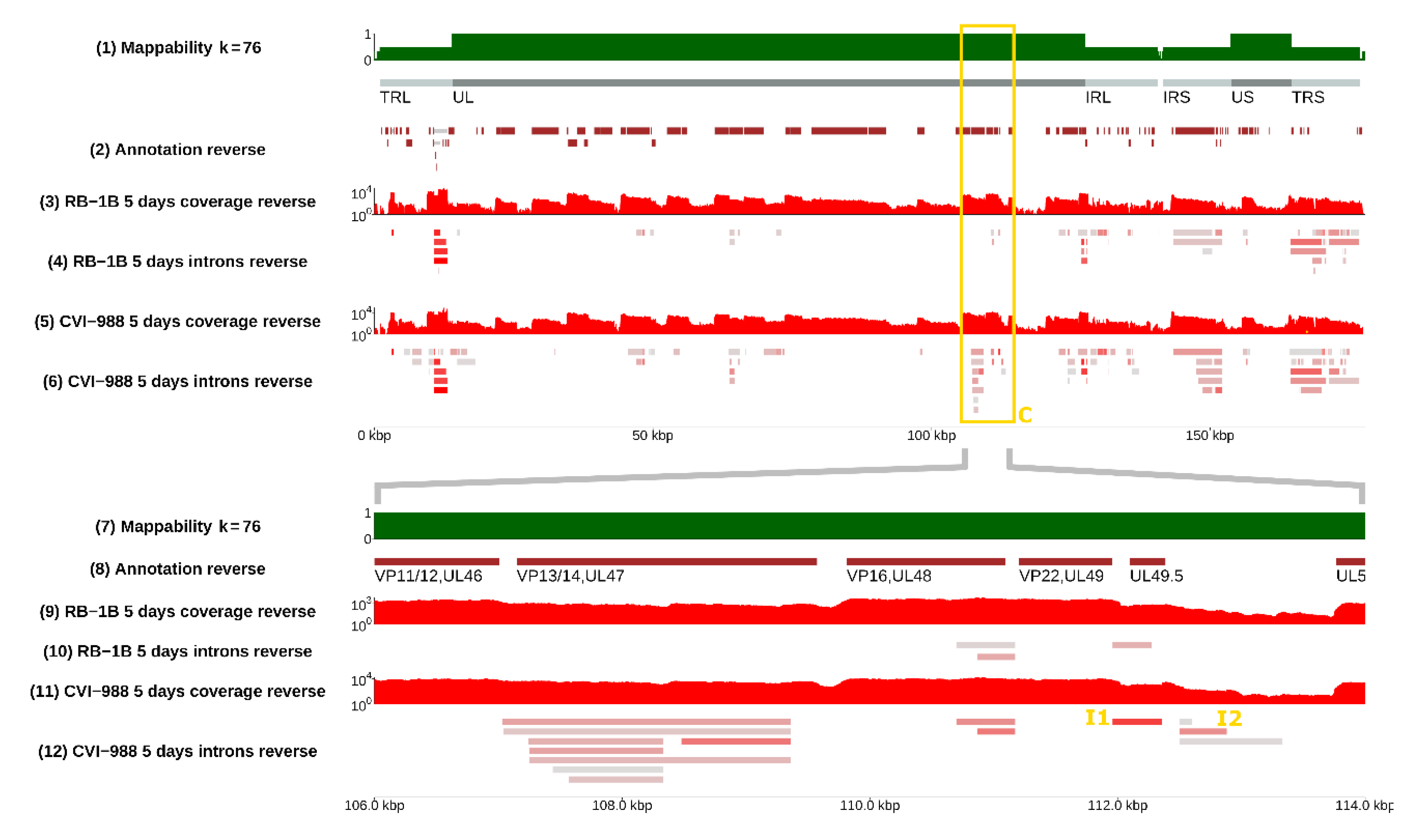
3.4. Selection of Strain-Dependent Introns
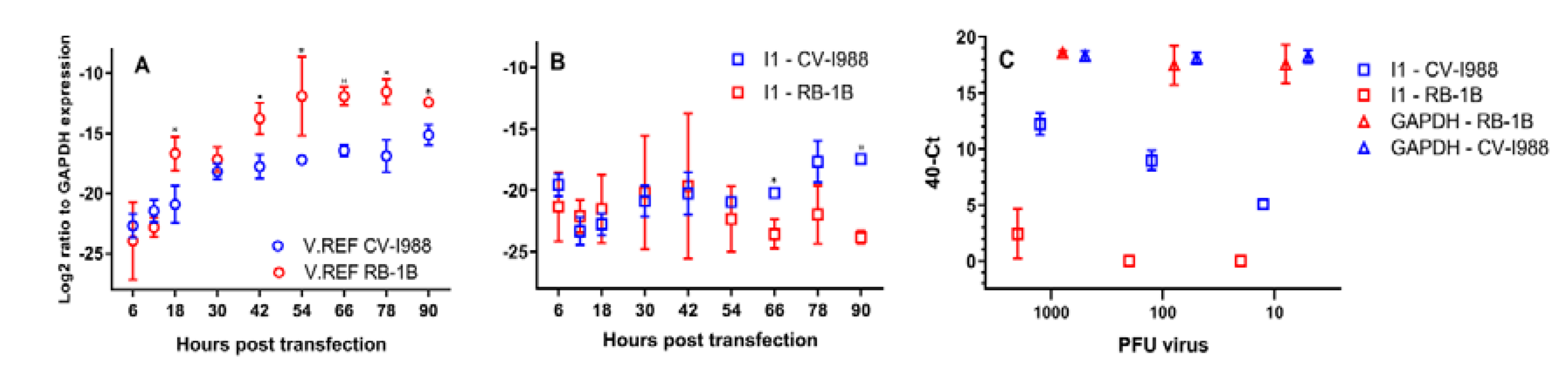
3.5. The Kinetics of I1 Expression in CEF Cells Transfected with CVI-988 and RB-1B
3.6. I1 Could Be Detected at a Very Low Level
- The expression of the housekeeping gene GAPDH remained unchanged at a 40-Ct value of approximately 18 across the different PFU levels (p = 0.48 on the basis of a two-way repeated measures ANOVA test) and independent of the strain.
- In CEF cells infected by CVI-988, the levels of expression of I1 correlated very well with inoculum titres (Pearson’s r = 0.98, p-value = 6.1 × 10−6), and I1 was detectable across the whole PFU range.
- I1 expression was undetectable in 1.3 × 106 CEF cells infected with 100 or 10 PFU of RB-1B. In cells infected with 1000 PFU, I1 levels were still extremely low (averaged 40-Ct value of 2.4).
3.7. Differential Expression of I1, I2, and I3 in RB-1B and CVI-988
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schat, K.; Nair, V. Neoplastic disease: Marek’s disease. In Diseases of Poultry, 13th ed.; Swayne, D., Glisson, J., McDougald, L., Nolan, L., Suarez, D., Nair, V., Eds.; Wyley-Blackwell: Hoboken, NJ, USA, 2013; pp. 515–552. [Google Scholar]
- Baigent, S.J.; Smith, L.P.; Nair, V.K.; Currie, R.J. Vaccinal control of Marek’s disease: Current challenges, and future strategies to maximize protection. Vet. Immunol. Immunopathol. 2006, 112, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.M.; Izumiya, Y.; Lupiani, B. Marek’s disease vaccines: Current status, and strategies for improvement and development of vector vaccines. Vet. Microbiol. 2017, 206, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Rispens, B.H.; van Vloten, H.; Mastenbroek, N.; Maas, J.L.; Schat, K.A. Control of Marek’s disease in the Netherlands. II. Field trials on vaccination with an avirulent strain (CVI 988) of Marek’s disease virus. Avian Dis. 1972, 16, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Rispens, B.H.; van Vloten, H.; Mastenbroek, N.; Maas, H.J.; Schat, K.A. Control of Marek’s disease in the Netherlands. I. Isolation of an avirulent Marek’s disease virus (strain CVI 988) and its use in laboratory vaccination trials. Avian Dis. 1972, 16, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.A.; Walkden-Brown, S.W.; Nair, V.K. Imperfect Vaccination Can Enhance the Transmission of Highly Virulent Pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef] [PubMed]
- Witter, R.L. Marek’s disease: The continuing struggle between pathogen and host. Vet. J. 2005, 170, 149–150. [Google Scholar] [CrossRef]
- Witter, R.L. The changing landscape of Marek’s disease. Avian Pathol 1998, 27, S46–S53. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Cairns, C.; Jones, M.J.; Bell, A.S.; Salathe, R.M.; Baigent, S.J.; Nair, V.K.; Dunn, P.A.; Read, A.F. Industry-Wide Surveillance of Marek’s Disease Virus on Commercial Poultry Farms. Avian Dis. 2017, 61, 153–164. [Google Scholar] [CrossRef]
- Baigent, S.J.; Smith, L.P.; Petherbridge, L.J.; Nair, V.K. Differential quantification of cloned CVI988 vaccine strain and virulent RB-1B strain of Marek’s disease viruses in chicken tissues, using real-time PCR. Res. Vet. Sci. 2011, 91, 167–174. [Google Scholar] [CrossRef]
- Baigent, S.J.; Nair, V.K.; Le Galludec, H. Real-time PCR for differential quantification of CVI988 vaccine virus and virulent strains of Marek’s disease virus. J. Virol. Methods 2016, 233, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Tombacz, D.; Balazs, Z.; Csabai, Z.; Moldovan, N.; Szucs, A.; Sharon, D.; Snyder, M.; Boldogkoi, Z. Characterization of the Dynamic Transcriptome of a Herpesvirus with Long-read Single Molecule Real-Time Sequencing. Sci. Rep. 2017, 7, 43751. [Google Scholar] [CrossRef] [PubMed]
- Tombacz, D.; Csabai, Z.; Szucs, A.; Balazs, Z.; Moldovan, N.; Sharon, D.; Snyder, M.; Boldogkoi, Z. Long-Read Isoform Sequencing Reveals a Hidden Complexity of the Transcriptional Landscape of Herpes Simplex Virus Type 1. Front. Microbiol. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Huertas, C.S.; Dominguez-Zotes, S.; Lechuga, L.M. Analysis of alternative splicing events for cancer diagnosis using a multiplexing nanophotonic biosensor. Sci. Rep. 2017, 7, 41368. [Google Scholar] [CrossRef]
- Sah, N.K.; Seniya, C. Survivin splice variants and their diagnostic significance. Tumour Biol. 2015, 36, 6623–6631. [Google Scholar] [CrossRef]
- Leman, R.; Gaildrat, P.; Le Gac, G.; Ka, C.; Fichou, Y.; Audrezet, M.P.; Caux-Moncoutier, V.; Caputo, S.M.; Boutry-Kryza, N.; Leone, M.; et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: An international collaborative effort. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nature Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedlander, M.R.; AC’t Hoen, P.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef]
- Derrien, T.; Estellé, J.; Marco Sola, S.; Knowles, D.G.; Raineri, E.; Guigó, R.; Ribeca, P. Fast computation and applications of genome mappability. PLoS One 2012, 7, e30377. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, M.E.; Uzilov, A.V.; Stein, L.D.; Mungall, C.J.; Holmes, I.H. JBrowse: A next-generation genome browser. Genome Res. 2009, 19, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baigent, S.J.; Petherbridge, L.J.; Howes, K.; Smith, L.P.; Currie, R.J.; Nair, V.K. Absolute quantitation of Marek’s disease virus genome copy number in chicken feather and lymphocyte samples using real-time PCR. J. Virol Methods 2005, 123, 53–64. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Moldovan, N.; Tombacz, D.; Szucs, A.; Csabai, Z.; Snyder, M.; Boldogkoi, Z. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol. 2017, 8, 2708. [Google Scholar] [CrossRef] [Green Version]
- Tombacz, D.; Csabai, Z.; Olah, P.; Balazs, Z.; Liko, I.; Zsigmond, L.; Sharon, D.; Snyder, M.; Boldogkoi, Z. Full-Length Isoform Sequencing Reveals Novel Transcripts and Substantial Transcriptional Overlaps in a Herpesvirus. PLoS ONE 2016, 11, e0162868. [Google Scholar] [CrossRef] [Green Version]
- O’Grady, T.; Cao, S.; Strong, M.J.; Concha, M.; Wang, X.; Splinter Bondurant, S.; Adams, M.; Baddoo, M.; Srivastav, S.K.; Lin, Z.; et al. Global bidirectional transcription of the Epstein-Barr virus genome during reactivation. J. Virol. 2014, 88, 1604–1616. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Shirazi, Y. Characterization of the protein product encoded by a splicing variant of the Marek’s disease virus Eco-Q gene (Meq). Virology 1996, 226, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Jarosinski, K.W.; Schat, K.A. Multiple alternative splicing to exons II and III of viral interleukin-8 (vIL-8) in the Marek’s disease virus genome: The importance of vIL-8 exon I. Virus Genes 2007, 34, 9–22. [Google Scholar] [CrossRef]
- Li, D.S.; Pastorek, J.; Zelnik, V.; Smith, G.D.; Ross, L.J. Identification of novel transcripts complementary to the Marek’s disease virus homologue of the ICP4 gene of herpes simplex virus. J. Gen. Virol. 1994, 75 (Pt. 7), 1713–1722. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Osterrieder, N. Marek’s disease virus expresses multiple UL44 (gC) variants through mRNA splicing that are all required for efficient horizontal transmission. J. Virol. 2012, 86, 7896–7906. [Google Scholar] [CrossRef] [Green Version]
- Schippers, T.; Jarosinski, K.; Osterrieder, N. The ORF012 gene of Marek’s disease virus type 1 produces a spliced transcript and encodes a novel nuclear phosphoprotein essential for virus growth. J. Virol. 2015, 89, 1348–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hearn, C.; Preeyanon, L.; Hunt, H.D.; York, I.A. An MHC class I immune evasion gene of Mareks disease virus. Virology 2015, 475, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Jarosinski, K.W.; Schat, K.A. Expression of Marek’s disease virus phosphorylated polypeptide pp38 produces splice variants and enhances metabolic activity. Vet. Microbiol. 2006, 117, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Rock, D.L.; Kutish, G.F. The genome of a very virulent Marek’s disease virus. J. Virol. 2000, 74, 7980–7988. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.F.; Wu, P.; Sui, D.; Ren, D.; Kamil, J.; Kung, H.J.; Witter, R.L. The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc. Natl. Acad. Sci. USA 2000, 97, 6091–6096. [Google Scholar] [CrossRef] [Green Version]
- Spatz, S.J.; Petherbridge, L.; Zhao, Y.; Nair, V. Comparative full-length sequence analysis of oncogenic and vaccine (Rispens) strains of Marek’s disease virus. J. Gen. Virol. 2007, 88, 1080–1096. [Google Scholar] [CrossRef]
- Hong, Y.; Coussens, P.M. Identification of an immediate-early gene in the Marek’s disease virus long internal repeat region which encodes a unique 14-kilodalton polypeptide. J. Virol. 1994, 68, 3593–3603. [Google Scholar] [CrossRef] [Green Version]
- Parcells, M.S.; Lin, S.F.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.R.; Chen, H.C.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D.; et al. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): Characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef] [Green Version]
- Petherbridge, L.; Howes, K.; Baigent, S.J.; Sacco, M.A.; Evans, S.; Osterrieder, N.; Nair, V. Replication-competent bacterial artificial chromosomes of Marek’s disease virus: Novel tools for generation of molecularly defined herpesvirus vaccines. J. Virol. 2003, 77, 8712–8718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petherbridge, L.; Brown, A.C.; Baigent, S.J.; Howes, K.; Sacco, M.A.; Osterrieder, N.; Nair, V.K. Oncogenicity of virulent Marek’s disease virus cloned as bacterial artificial chromosomes. J. Virol. 2004, 78, 13376–13380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, T.F.; Nair, V.; Institute for Animal Health (Great Britain). Marek’s Disease: An Evolving Problem; Elsevier: Amesterdam, The Netherlands, 2004. [Google Scholar]
- Tirosh, O.; Cohen, Y.; Shitrit, A.; Shani, O.; Le-Trilling, V.T.; Trilling, M.; Friedlander, G.; Tanenbaum, M.; Stern-Ginossar, N. The Transcription and Translation Landscapes during Human Cytomegalovirus Infection Reveal Novel Host-Pathogen Interactions. PLoS Pathog. 2015, 11, e1005288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armero, V.E.S.; Tremblay, M.P.; Allaire, A.; Boudreault, S.; Martenon-Brodeur, C.; Duval, C.; Durand, M.; Lapointe, E.; Thibault, P.; Tremblay-Letourneau, M.; et al. Transcriptome-wide analysis of alternative RNA splicing events in Epstein-Barr virus-associated gastric carcinomas. PLoS ONE 2017, 12, e0176880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertzbach, L.D.; Pfaff, F.; Pauker, V.I.; Kheimar, A.M.; Hoper, D.; Hartle, S.; Karger, A.; Kaufer, B.B. The Transcriptional Landscape of Marek’s Disease Virus in Primary Chicken B Cells Reveals Novel Splice Variants and Genes. Viruses 2019, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Schat, K.A.; Calnek, B.W.; Fabricant, J. Characterisation of two highly oncogenic strains of Marek’s disease virus. Avian Pathol. 1982, 11, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Conradie, A.M.; Bertzbach, L.D.; Bhandari, N.; Parcells, M.; Kaufer, B.B. A Common Live-Attenuated Avian Herpesvirus Vaccine Expresses a Very Potent Oncogene. mSphere 2019, 4, e00658-19. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MDV Strain | Reads Mapping to the Forward Viral Strand | Alignments to the Forward Viral Strand | Reads Mapping to the Reverse Viral Strand | Alignments to the Reverse Viral Strand |
|---|---|---|---|---|
| CVI-988 replicate 1 | 949,845 | 1,273,630 | 639,578 | 966,599 |
| CVI-988 replicate 2 | 1,391,374 | 1,865,743 | 932,231 | 1,404,683 |
| RB-1B replicate 1 | 717,633 | 1,213,552 | 549,656 | 998,707 |
| RB-1B replicate 2 | 650,431 | 1,110,938 | 496,594 | 914,609 |
| Genomic Region | Spliceforms In CVI-988 | Spliceforms In RB-1B | Function |
|---|---|---|---|
| vIL8 | 5 | 5 | Viral IL8 |
| pp14A/B | 6 | 5 | 14KD lytic proteins |
| Lip | 3 | 2 | Lipase |
| UL3 | 5 | 3 | Nuclear phosphoprotein |
| UL15 | 17 | 4 | DNA packaging protein |
| UL19 | 4 | 4 | Major capsid protein |
| UL21 | 3 | 2 | Tegument protein |
| UL24 | 2 | 2 | Reactivating from latency/immune evasion |
| UL28 | 7 | 1 | DNA packaging protein. |
| UL29 | 2 | 1 | DNA binding protein |
| UL34 | 2 | 1 | Membrane phosphoprotein |
| UL38 | 2 | 1 | Capsid protein |
| UL41 | 4 | 1 | Tegument protein |
| UL44 | 6 | 5 | Glycoprotein C |
| UL46 | 2 | 1 | Tegument phosphoprotein |
| UL47 | 11 | 1 | Tegument phosphoprotein |
| UL48 | 3 | 3 | Tegument immediate-early protein |
| UL49 | 2 | 2 | Tegument phosphoprotein |
| UL52/UL53/UL54 | 14 | 4 | DNA helicase/glycoprotein K/ICP27-like proteins |
| RLORF14a | 3 | 3 | 38 KD protein |
| ICP4 | 5 | 1 | IE protein |
| ICP4 area | 15 | 7 | - |
| US7 | 5 | 3 | Glycoprotein I |
| US8 | 3 | 3 | Glycoprotein E |
| MDV Strain | Strand | Position of First Intron Nucleotide | Position of Last Intron Nucleotide | Gene | Read Coverage | Name |
|---|---|---|---|---|---|---|
| A. Introns only spliced in MDV strain CVI-988 | ||||||
| GaHV2 | − | 112359 | 111959 | VP22 gene, intergenic region | 646 | I1 |
| GaHV2 | − | 112881 | 112500 | UL50 | 58 | I2 |
| GaHV2 | + | 43021 | 43723 | UL15, terminase | 39 | I3 |
| GaHV2 | + | 43021 | 43166 | UL15, terminase | 42 | - |
| GaHV2 | + | 50934 | 51306 | UL21 | 38 | |
| GaHV2 | − | 108334 | 107252 | UL46/UL47 | 29 | |
| GaHV2 | + | 43021 | 43808 | UL15, terminase | 28 | |
| B. Introns only spliced in MDV strain RB-1B | ||||||
| GaHV2 | − | 111959 | 112277 | UL49/UL49.5 | 19 | - |
| C. The intron spliced in cells infected with both of the strains | ||||||
| GaHV2 | − | 170198 | 170114 | ICP4/LAT | 183 | V.Ref |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadigh, Y.; Tahiri-Alaoui, A.; Spatz, S.; Nair, V.; Ribeca, P. Pervasive Differential Splicing in Marek’s Disease Virus Can Discriminate CVI-988 Vaccine Strain from RB-1B Very Virulent Strain in Chicken Embryonic Fibroblasts. Viruses 2020, 12, 329. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030329
Sadigh Y, Tahiri-Alaoui A, Spatz S, Nair V, Ribeca P. Pervasive Differential Splicing in Marek’s Disease Virus Can Discriminate CVI-988 Vaccine Strain from RB-1B Very Virulent Strain in Chicken Embryonic Fibroblasts. Viruses. 2020; 12(3):329. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030329
Chicago/Turabian StyleSadigh, Yashar, Abdessamad Tahiri-Alaoui, Stephen Spatz, Venugopal Nair, and Paolo Ribeca. 2020. "Pervasive Differential Splicing in Marek’s Disease Virus Can Discriminate CVI-988 Vaccine Strain from RB-1B Very Virulent Strain in Chicken Embryonic Fibroblasts" Viruses 12, no. 3: 329. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030329