Development and Application of Performance Assessment Criteria for Next-Generation Sequencing-Based HIV Drug Resistance Assays


Abstract
:1. Introduction
2. Materials and Methods
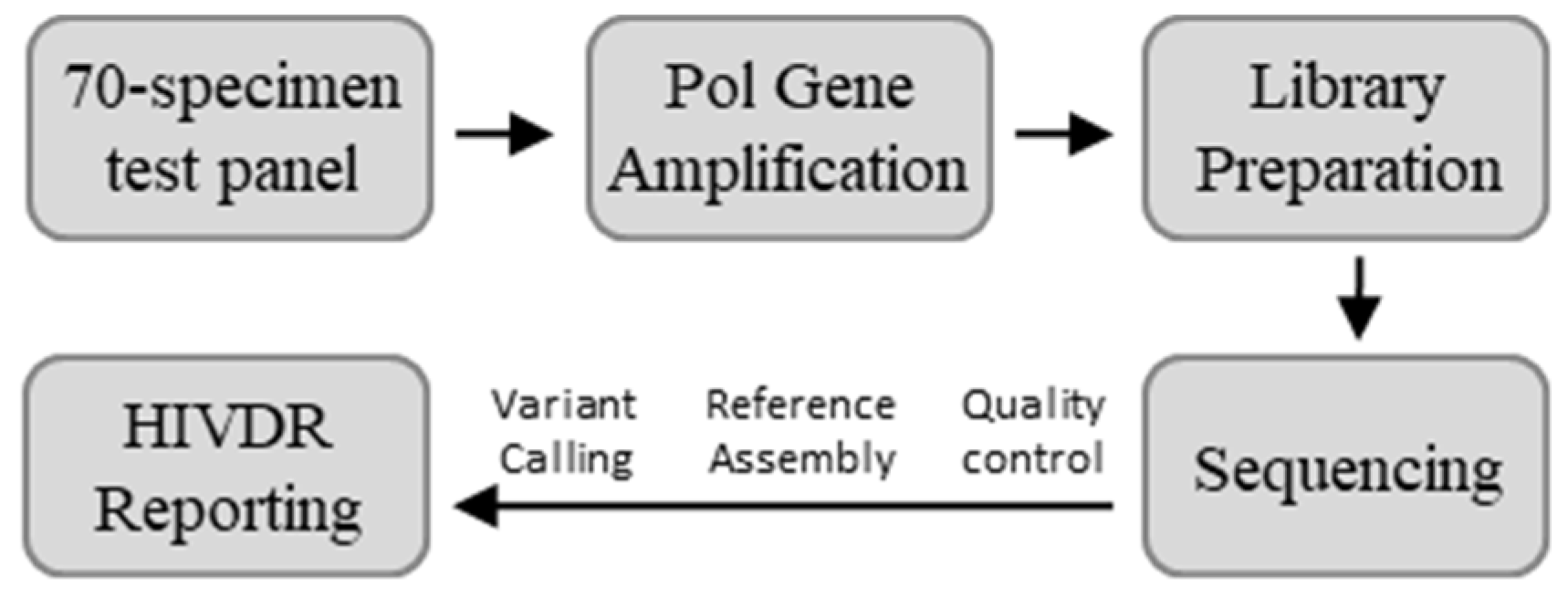
2.1. Specimens
2.2. NGS-Based HIVDR Assays
2.3. Selection of Assessment Criteria for NGS-Based HIVDR Assays
2.4. Assessment of NGS HIVDR Assays with Each of the Proposed Criteria
3. Results
3.1. Determining the Lower Boundary of the DRM Measuring Interval
3.2. Linear Range for the DRM Detection
3.3. Accuracy and Precision of the HIVDR Assays
3.4. Assay Error, Analytical Specificity, and Analytical Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). WHO Global Action Plan on HIV Drug Resistance 2017–2021. Available online: https://www.who.int/hiv/pub/drugresistance/hivdr-action-plan-2017-2021/en/ (accessed on 10 March 2020).
- Günthard, H.F.; Calvez, V.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Jacobsen, D.M.; Richman, D.D. Human Immunodeficiency Virus Drug Resistance: 2018 Recommendations of the International Antiviral Society–USA Panel. Clin. Infect. Dis. 2019, 68, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Derache, A.; Iwuji, C.C.; Baisley, K.; Danaviah, S.; Marcelin, A.G.; Calvez, V.; De Oliveira, T.; Dabis, F.; Porter, K.; Pillay, D. Impact of next-generation sequencing defined human immunodeficiency virus pretreatment drug resistance on virological outcomes in the ANRS 12249 treatment-as-prevention trial. Clin. Infect. Dis. 2019, 69, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inzaule, S.C.; Hamers, R.L.; Noguera-Julian, M.; Casadellà, M.; Parera, M.; Kityo, C.; Steegen, K.; Naniche, D.; Clotet, B.; Rinke de Wit, T.F.; et al. Clinically relevant thresholds for ultrasensitive HIV drug resistance testing: A multi-country nested case-control study. Lancet HIV 2018, 5, e638–e646. [Google Scholar] [CrossRef]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; Macarthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-Abundance Drug-Resistant Viral Variants in Chronically HIV-Infected, Antiretroviral Treatment-Naive Patients Significantly Impact Treatment Outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Ríos, S.; García-Morales, C.; Matías-Florentino, M.; Romero-Mora, K.A.; Tapia-Trejo, D.; Quiroz-Morales, V.S.; Reyes-Gopar, H.; Ji, H.; Sandstrom, P.; Casillas-Rodríguez, J.; et al. Pretreatment HIV-drug resistance in Mexico and its impact on the effectiveness of first-line antiretroviral therapy: A nationally representative 2015 WHO survey. Lancet HIV 2016, 3, e579–e591. [Google Scholar] [CrossRef]
- Gibson, R.M.; Schmotzer, C.L.; Quiñones-Mateu, M.E. Next-generation sequencing to help monitor patients infected with HIV: Ready for clinical use? Curr. Infect. Dis. Rep. 2014, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- WHO. HIV Drug Resistance Laboratory Training Package; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Burd, E.M. Validation of laboratory-developed molecular assays for infectious diseases. Clin. Microbiol. Rev. 2010, 23, 550–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, R.J.; Phan, K.L.; Saboor, A.; Miller, J.D.; Muehlenbachs, A. Practical Guidance to Implementing Quality Management Systems in Public Health Laboratories Performing Next-Generation Sequencing: Personnel, Equipment, and Process Management (Phase 1). J. Clin. Microbiol. 2019, 57, e00261-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.R.; Gao, F.; Sandstrom, P.; Ji, H. External quality assessment for next-generation sequencing-based HIV drug resistance testing: Unique requirements and challenges. Viruses 2020, 12, 550. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.; Lee, E.R.; Nykoluk, M.; Enns, E.; Liang, B.; Capina, R.; Gauthier, M.K.; Domselaar, G.V.; Sandstrom, P.; Brooks, J.; et al. A MiSeq-HyDRA platform for enhanced HIV drug resistance genotyping and surveillance. Sci. Rep. 2019, 9, 8970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Lubin, I.M. Assuring the quality of NGS in clinical lab practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.; Van Deerlin, V.M.; Gulley, M.L. Recommended principles and practices for validating clinical molecular pathology tests. Arch. Pathol. Lab. Med. 2009, 133, 743–755. [Google Scholar] [PubMed]
- Cottrell, C.E.; Al-Kateb, H.; Bredemeyer, A.J.; Duncavage, E.J.; Spencer, D.H.; Abel, H.J.; Lockwood, C.M.; Hagemann, I.S.; O’Guin, S.M.; Burcea, L.C.; et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J. Mol. Diagn. 2014, 16, 89–105. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Manual for Organizing a National External Quality Assessment Programme for Health Laboratories and Other Testing Sites. 2016. Available online: https://www.who.int/hiv/pub/toolkits/manual-external-quality-assessment-testing/en/ (accessed on 20 April 2020).
- Norman, K.L.; Dinauer, D.M. Practices of Sequencing Quality Assurance. In Molecular Microbiology: Diagnostic Principles and Practice; Persing, D.H., Ed.; John Wiley & Sons: Washington, DC, USA, 2016; pp. 766–783. ISBN 9781555819071. [Google Scholar]
- CLSI document MM09A2: Nucleic Acid Sequencing Methods in Diagnostic Laboratory Medicine. In Approved Guideline, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2014.
- Miller, W.G.; Jones, G.R.D.; Horowitz, G.L.; Weykamp, C. Proficiency testing/external quality assessment: Current challenges and future directions. Clin. Chem. 2011, 57, 1670–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorsson, E. Validation and verification of measurement methods in clinical chemistry. Bioanalysis 2012, 4, 305–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Enns, E.; Brumme, C.J.; Parkin, N.; Howison, M.; Lee, E.R.; Capina, R.; Marinier, E.; Avila-Rios, S.; Sandstrom, P.; et al. Bioinformatic data processing pipelines in support of next-generation sequencing-based HIV drug resistance testing: The Winnipeg Consensus. J. Int. AIDS Soc. 2018, 21, e25193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef] [PubMed]
- CLSI Document EP17-A2: Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012.
- Paredes, R.; Lalama, C.M.; Ribaudo, H.J.; Schackman, B.R.; Shikuma, C.; Giguel, F.; Iii, W.A.M.; Johnson, V.A.; Fiscus, S.A.; D’aquila, R.T.; et al. Pre-existing Minority Drug-Resistant HIV-1 Variants, Adherence, and Risk of Antiretroviral Treatment Failure. J. Infect. Dis. 2010, 201, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Finney, D.J.; Stevens, W.L. A table for the calculation of working probits and weights in probit analysis. Biometrika 1948, 35, 191–201. [Google Scholar] [CrossRef] [PubMed]
- CLSI Document EP06-A: Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical Approach, 1st ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2003.
- CLSI document EP05-A2: Evaluation of Precision Performance of Quantitative Measurement Methods. In Approved Guideline, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2005.
- Hatzakis, A.; Papachristou, H.; Nair, S.J.; Fortunko, J.; Foote, T.; Kim, H.; Peling, T.L.; Worlock, A.J. Analytical characteristics and comparative evaluation of Aptima HIV-1 Quant Dx assay with Ampliprep/COBAS TaqMan HIV-1 test v2.0. Virol. J. 2016, 13, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.V.; Kim, H.C.; Fortunko, J.; Foote, T.; Peling, T.; Tran, C.; Nugent, C.T.; Joo, S.; Kang, Y.; Wilkins, B.; et al. Aptima HIV-1 Quant Dx-A fully automated assay for both diagnosis and quantification of HIV-1. J. Clin. Virol. 2016, 77, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Sandstrom, P.; Paredes, R.; Harrigan, P.R.; Brumme, C.J.; Avila-Rios, S.; Kantor, R. Are we ready for NGS HIV drug resistance testing? The second Winnipeg symposium. Viruses 2020, 12, 586. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Braverman, M.S.; Abbate, I.; Aerssens, J.; Bidet, Y.; Bouchez, O.; Gabriel, C.; Izopet, J.; Kessler, H.H.; Stelzl, E.; et al. An international multicenter study on HIV-1 drug resistance testing by 454 ultra-deep pyrosequencing. J. Virol. Methods 2014, 204, 31–37. [Google Scholar] [CrossRef] [PubMed]
- St John, E.P.; Simen, B.B.; Turenchalk, G.S.; Braverman, M.S.; Abbate, I.; Aerssens, J.; Bouchez, O.; Gabriel, C.; Izopet, J.; Meixenberger, K.; et al. A follow-up of the multicenter collaborative study on HIV-1 drug resistance and tropism testing using 454 ultra deep pyrosequencing. PLoS ONE 2016, 11, e0146687. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadellà, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance comparison of next generation sequencing analysis pipelines for HIV-1 drug resistance testing. Sci. Rep. 2020, 10, 1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. World Health Organization Global Strategy for the Surveillance and Monitoring or HIV Drug Resistance. 2012. Available online: https://www.who.int/hiv/pub/drugresistance/drug_resistance_strategy/en/ (accessed on 27 May 2020).
- Eisinger, R.W.; Dieffenbach, C.W.; Fauci, A.S. HIV viral load and transmissibility of HIV infection undetectable equals untransmittable. J. Am. Med. Assoc. 2019, 321, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Saag, M.S.; Holodniy, M.; Kuritzkes, D.R.; O’Brien, W.A.; Coombs, R.; Poscher, M.E.; Jacobsen, D.M.; Shaw, G.M.; Richman, D.D.; Volberding, P.A. HIV viral load markers in clinical practice. Nat. Med. 1996, 2, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Next-Generation Human Immunodeficiency Virus Sequencing for Patient Management and Drug Resistance Surveillance. J. Infect. Dis. 2017, 216, S829–S833. [Google Scholar] [CrossRef] [PubMed]
- CLSI document EP15-A2: User Verification of Performance for Precision and Trueness. In Approved Guideline, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2006.
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Gondola, J.; Ortiz, A.Y.; Castillo, J.A.; Pascale, J.M.; Martinez, A.A. Barcoding analysis of HIV drug resistance mutations using Oxford Nanopore MinION (ONT) sequencing. bioRxiv 2018, 240077. [Google Scholar]
- Dessilly, G.; Goeminne, L.; Vandenbroucke, A.T.; Dufrasne, F.E.; Martin, A.; Kabamba-Mukabi, B. First evaluation of the next-generation sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLoS ONE 2018, 13, e0209561. [Google Scholar] [CrossRef] [PubMed]
- Maljkovic Berry, I.; Melendrez, M.C.; Bishop-Lilly, K.A.; Rutvisuttinunt, W.; Pollett, S.; Talundzic, E.; Morton, L.; Jarman, R.G. Next Generation Sequencing and Bioinformatics Methodologies for Infectious Disease Research and Public Health: Approaches, Applications, and Considerations for Development of Laboratory Capacity. J. Infect. Dis. 2019, 221, S292–S307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Li, D.; Xia, S.; Song, H.; Dong, Z.; Chen, F.; Sun, X.; Tang, Z. Absolute quantification of plasmid DNA by real-time PCR with genomic DNA as external standard and its application to a biodistribution study of an HIV DNA vaccine. Anal. Sci. 2009, 25, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, C.; Cromer, D.; Grimm, A.; Chopra, A.; Mallal, S.; Davenport, M.; Mak, J. A general method to eliminate laboratory induced recombinants during massive, parallel sequencing of cDNA library. Virol. J. 2015, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svarovskaia, E.S.; Delviks, K.A.; Hwang, C.K.; Pathak, V.K. Structural Determinants of Murine Leukemia Virus Reverse Transcriptase That Affect the Frequency of Template Switching. J. Virol. 2000, 74, 7171–7178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Leggett, R.M. Alvis: A tool for contig and read ALignment VISualisation and chimera detection. bioRxiv 2019, 663401. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.A.; Geretti, A.M. Low-frequency HIV-1 drug resistance mutations can be clinically significant but must be interpreted with caution. J. Antimicrob. Chemother. 2010, 65, 1322–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugelman, J.R.; Wiley, M.R.; Nagle, E.R.; Reyes, D.; Pfeffer, B.P.; Kuhn, J.H.; Sanchez-Lockhart, M.; Palacios, G.F. Error baseline rates of five sample preparation methods used to characterize RNA virus populations. PLoS ONE 2017, 12, e0171333. [Google Scholar] [CrossRef] [PubMed]
- Santiago, G.A.; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J.L. Analytical and Clinical Performance of the CDC Real Time RT-PCR Assay for Detection and Typing of Dengue Virus. PLoS Negl. Trop. Dis. 2013, 7, 36–38. [Google Scholar] [CrossRef]
- Schibler, M.; Yerly, S.; Vieille, G.; Docquier, M.; Turin, L.; Kaiser, L.; Tapparel, C. Critical Analysis of Rhinovirus RNA Load Quantification by Real-Time Reverse Transcription-PCR. J. Clin. Microbiol. 2012, 50, 2868–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. WHO Information for the Molecular Detection of Influenza Viruses. 2020. Available online: https://www.who.int/influenza/gisrs_laboratory/molecular_diagnosis/en/ (accessed on 27 May 2020).
- Li, J.Z.; Kuritzkes, D.R. Clinical Implications of HIV-1 Minority Variants. Clin. Infect. Dis. 2013, 56, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbunkah, H.A.; Bertagnolio, S.; Hamers, R.L.; Hunt, G.; Inzaule, S.; Rinke De Wit, T.F.; Paredes, R.; Parkin, N.T.; Jordan, M.R.; Metzner, K.J.; et al. Low-Abundance Drug-Resistant HIV-1 Variants in Antiretroviral Drug-Naive Individuals: A Systematic Review of Detection Methods, Prevalence, and Clinical Impact. J. Infect. Dis. 2019, 221, 1584–1597. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.; Lam, H.; Duan, H.; Ye, L.; Jongkam, N.; Chen, W.; Zhang, S.; Li, S. Benefits and Challenges with Applying Unique Molecular Identifiers in Next Generation Sequencing to Detect Low Frequency Mutations. PLoS ONE 2016, 11, e0146638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keys, J.R.; Zhou, S.; Anderson, J.A.; Eron, J.J.; Rackoff, L.A.; Jabara, C.; Swanstrom, R. Primer ID informs next-generation sequencing platforms and reveals preexisting drug resistance mutations in the HIV-1 reverse transcriptase coding domain. AIDS Res. Hum. Retrovir. 2015, 31, 658–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coefficient of Variation (CV, %) | ||||
|---|---|---|---|---|
| % DRMs | Intra-Run | Inter-Run | Inter-Operator | Inter-Lab |
| 1 | 12.7 | 5.2 | 2.8 | 15.1 |
| 2 | 6.9 | 10.3 | 1.7 | 15.9 |
| 5 | 7.7 | 5.3 | 3.0 | 13.1 |
| 10 | 6.2 | 4.6 | 1.6 | 15.7 |
| 20 | 8.2 | 7.2 | 5.9 | 7.7 |
| 100 | 0.1 | 0.3 | 0.2 | 0.1 |
| Performance Characteristic | Proposed Definition for NGS-Based HIVDR | Suggested Benchmark | References |
|---|---|---|---|
| DRM Measuring Interval | The range of DRMs that can be detected with an acceptable linearity, sensitivity, and precision. | 2–100% | [8,9,15] |
| Linear Range | The percentile range of DRM frequencies wherein the linear correlation is maintained between expected and observed values. | 2–100% | [9,15,18,27,39] |
| Precision | The extent to which repeated testing on identical samples renders comparable results with acceptable repeatability and reproducibility. | CV < 25% (DRMs < 50%) CV < 15% (DRMs ≥ 50%) | [9,15,28,40] |
| Accuracy | The extent to which the detected DRM frequency is in agreement with reference materials. The value is relative to the theoretical DRM frequency. | Error < 40% | [9,15,40] |
| Sequence Error Rate | The overall error in amino acid frequencies due to PCR, sequencing, and data processing steps. | <2% | [8,36,41] |
| Analytical Sensitivity | The probability that the assay detects a DRM within the measuring interval when it is present (measured as 1—false negative rate). | >99% (plasmid) | [8,9,13,15,24] |
| Analytical Specificity | The probability that the assay does not detect a DRM when it is absent (measured as 1—false positive rate). | >95% | [9,13,15] |
| Limit of the Viral Load | The lowest viral load level at which the test can still effectively detect DRMs from a sample. | 1000 copies/mL | [8,15,36] |
| Robustness | The capability of the assay to meet the above criteria using clinical samples of any major HIV subtype(s). | Coverage of all major subtypes | [8,13,15,36] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, M.G.; Liang, D.; Cooper, B.; Le, Y.; Taylor, T.; Lee, E.R.; Wu, S.; Sandstrom, P.; Ji, H. Development and Application of Performance Assessment Criteria for Next-Generation Sequencing-Based HIV Drug Resistance Assays. Viruses 2020, 12, 627. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060627
Becker MG, Liang D, Cooper B, Le Y, Taylor T, Lee ER, Wu S, Sandstrom P, Ji H. Development and Application of Performance Assessment Criteria for Next-Generation Sequencing-Based HIV Drug Resistance Assays. Viruses. 2020; 12(6):627. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060627
Chicago/Turabian StyleBecker, Michael G., Dun Liang, Breanna Cooper, Yan Le, Tracy Taylor, Emma R. Lee, Sutan Wu, Paul Sandstrom, and Hezhao Ji. 2020. "Development and Application of Performance Assessment Criteria for Next-Generation Sequencing-Based HIV Drug Resistance Assays" Viruses 12, no. 6: 627. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060627