Metatranscriptomic Identification of Diverse and Divergent RNA Viruses in Green and Chlorarachniophyte Algae Cultures

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Algal Cultures
2.2. Total RNA Extractions
2.3. Total RNA Sequencing
2.4. In Silico Processing of Meta-Transcriptomic Data
2.4.1. Read Depletion and Contig Assembly
2.4.2. RNA Virus Detection Using BLASTx and BLASTn
2.4.3. RNA Virus Profile-Based Homology Detection
2.4.4. D Protein Structure Prediction of RdRp-Like Contigs
2.4.5. RNA Virus Sequence Analysis and Annotation
2.4.6. Revealing Host-Virus Associations
2.4.7. Phylogenetic Analysis
2.5. RT-PCR Validation
2.6. Data Availability
3. Results
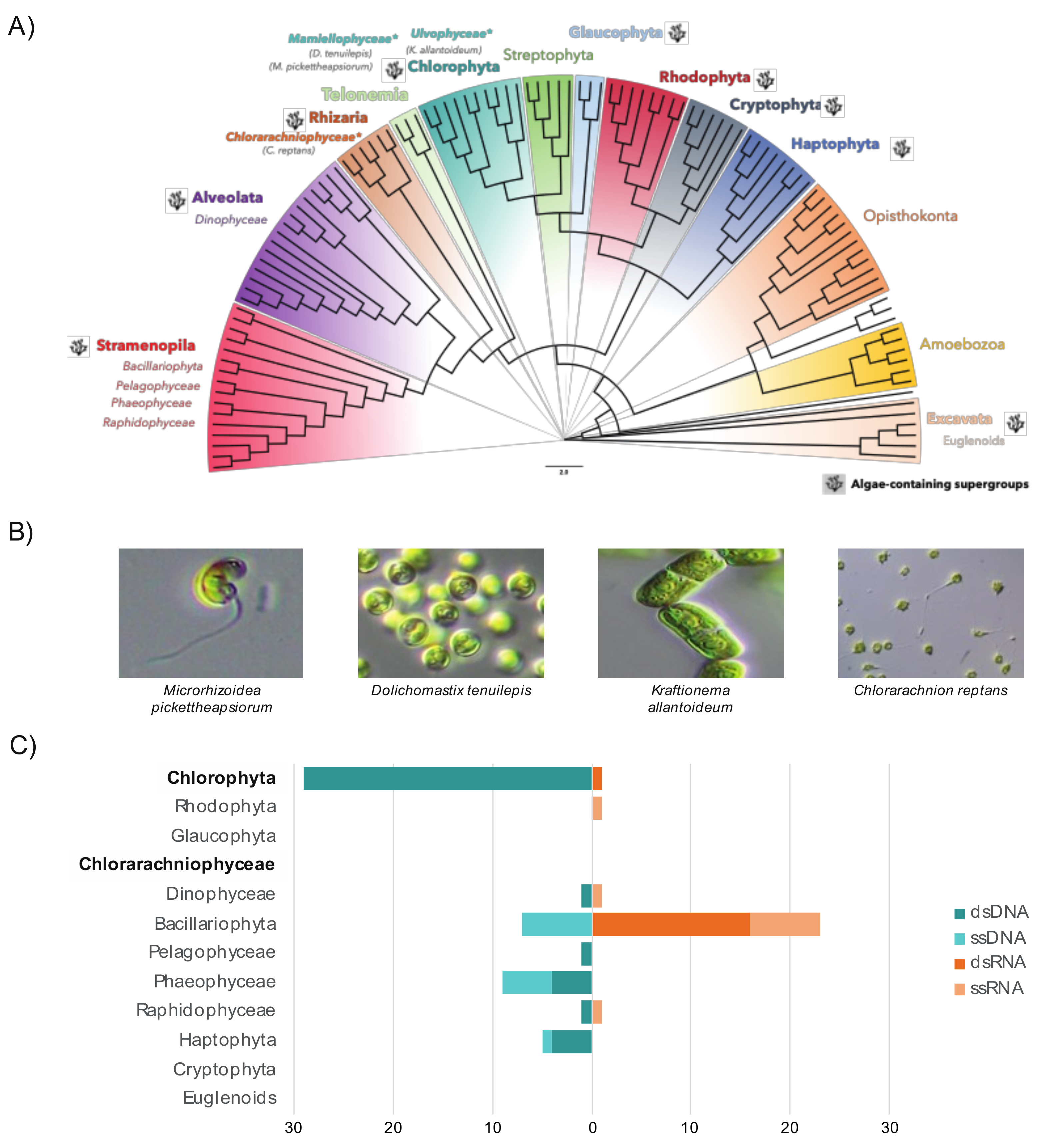
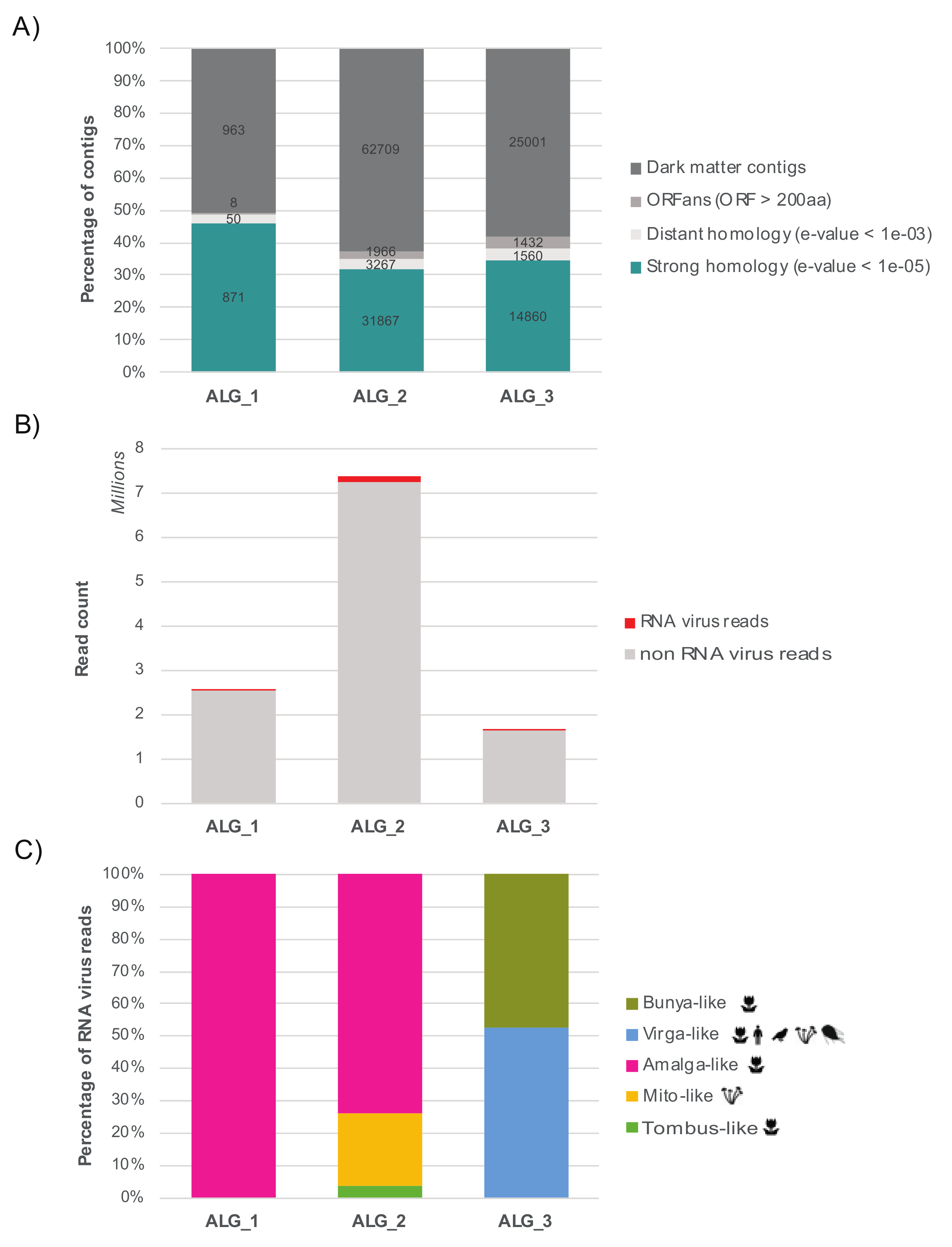
3.1. The RNA Viromes of Two Divergent Groups of Microalgae
3.2. Detection of Divergent Viruses Using Protein Structural Data
3.3. Relative Abundance and Prevalence of RNA Viruses in the Samples
3.4. Detection of Possible Secondary Hosts
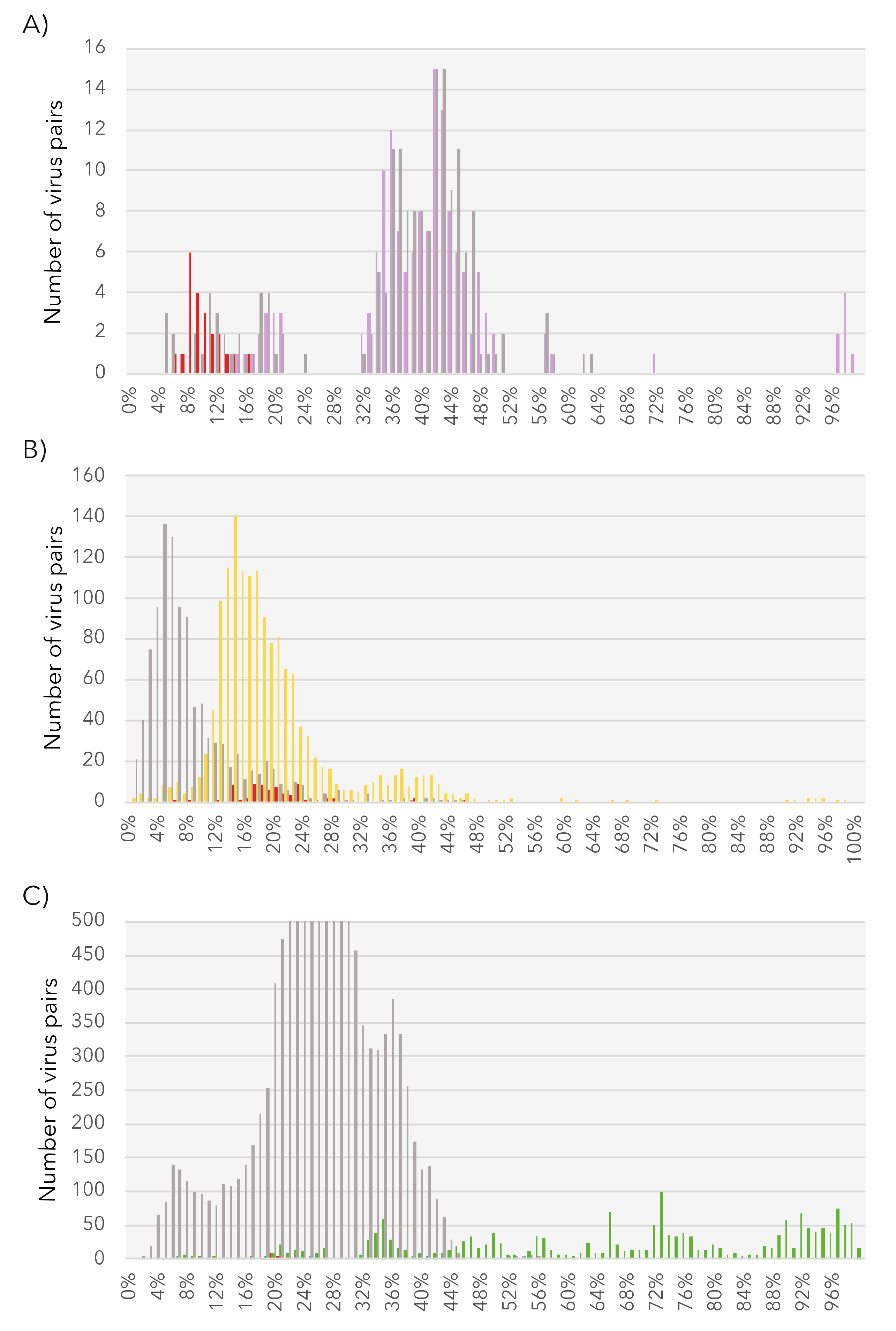
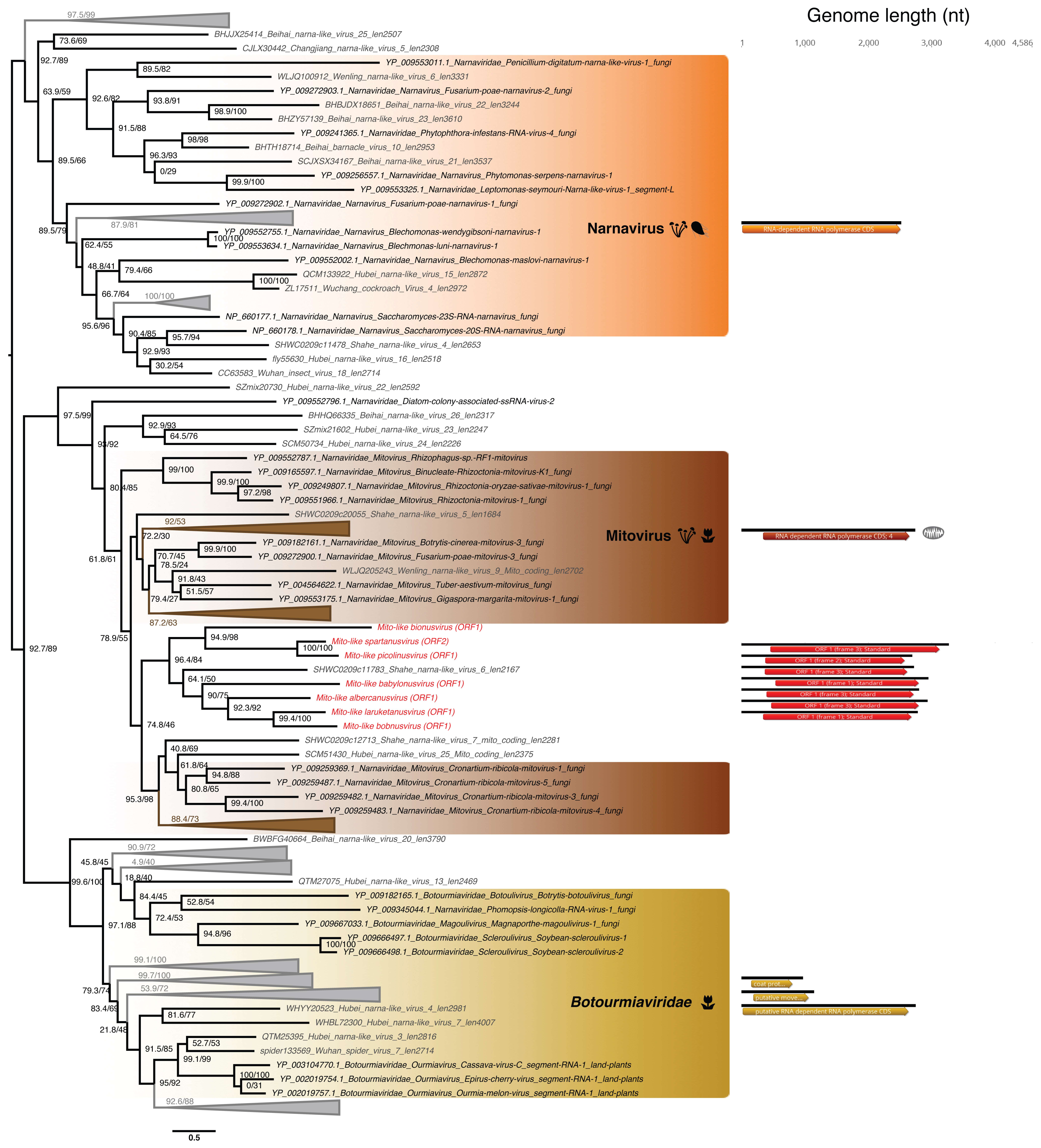
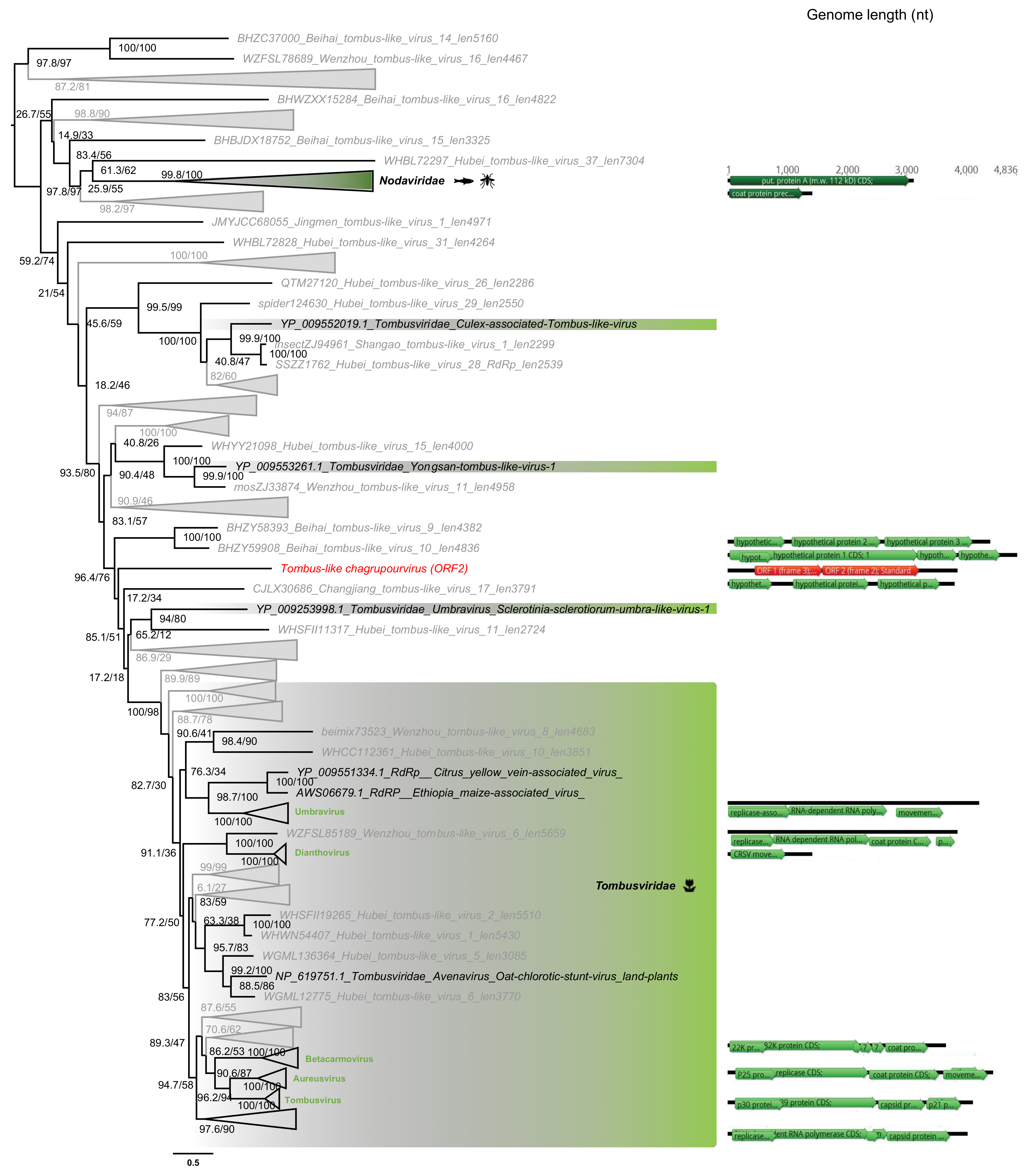
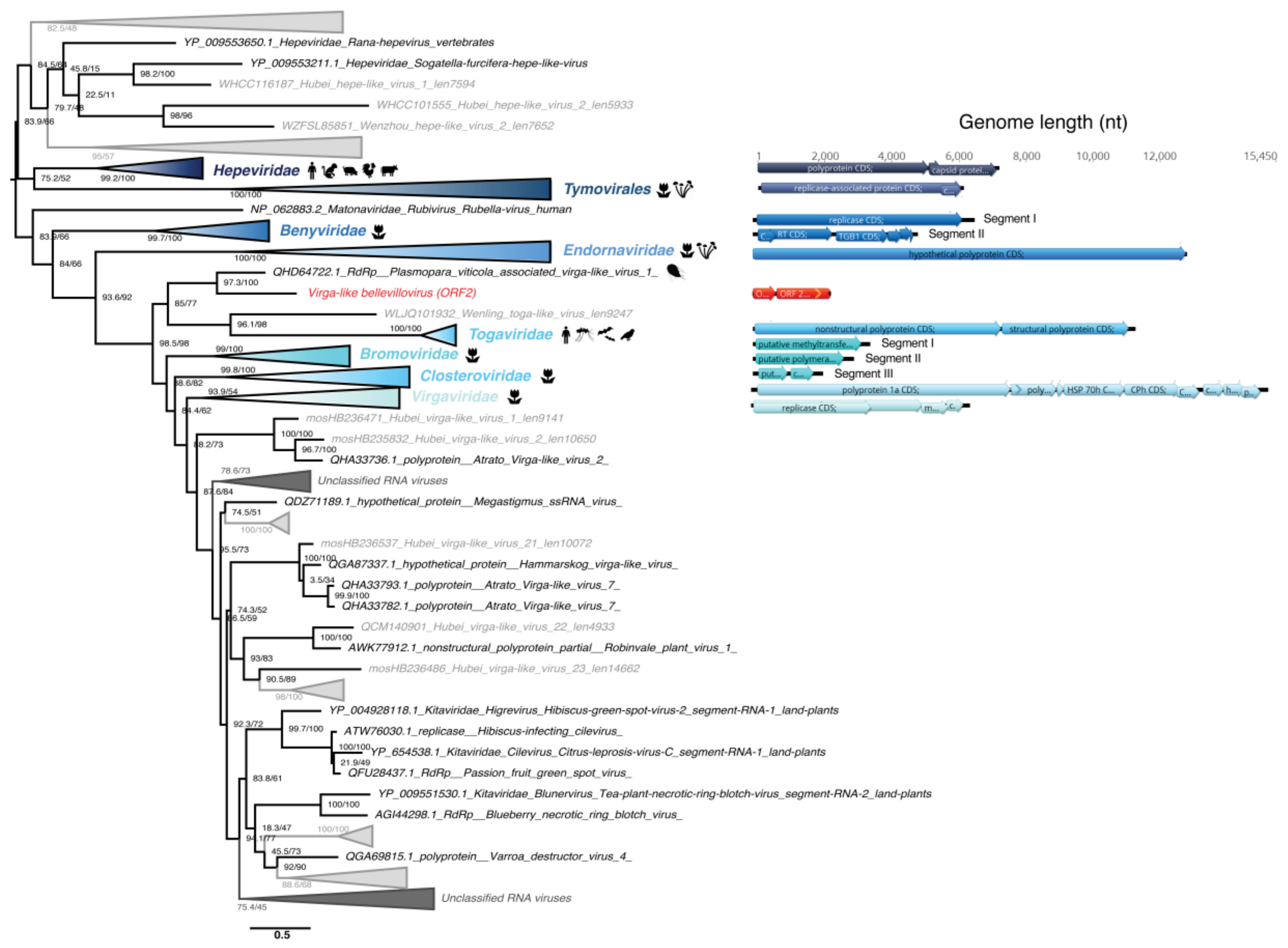
3.5. Phylogenetic Analysis of the Newly Identified Viruses
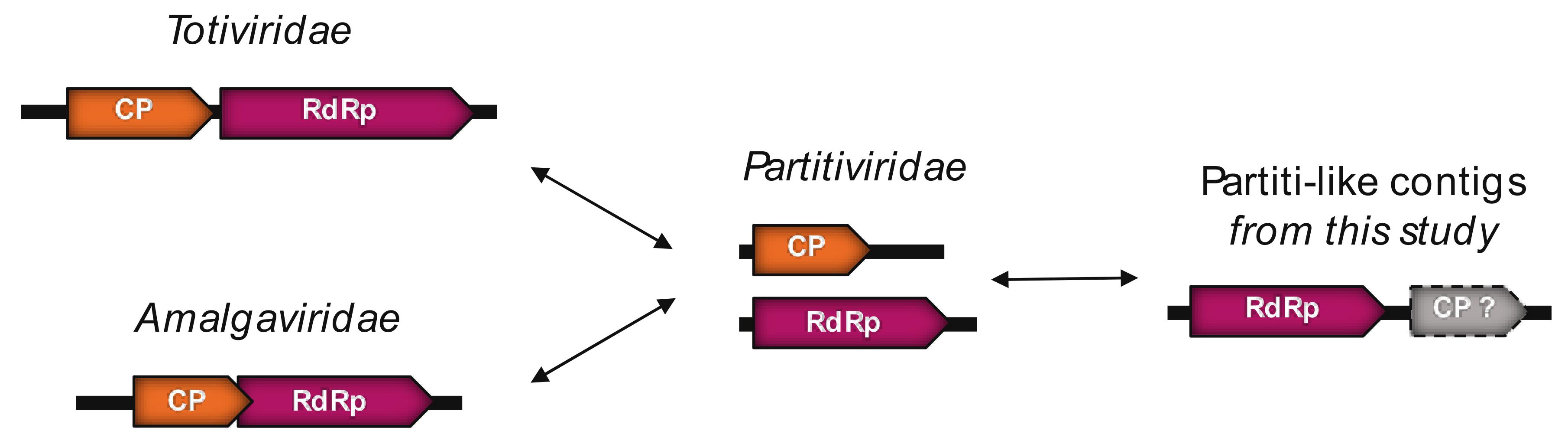
3.5.1. Partiti-Like dsRNA Viruses
3.5.2. Mitovirus-Like ssRNA(+) Viruses
3.5.3. Tombusviridae-Like ss(+)RNA Viruses
3.5.4. Virgaviridae-Like ssRNA(+) Viruses
3.5.5. Bunyavirales-Like ss(-)RNA Viruses
4. Discussion
4.1. RNA Virome Similarities between Green Algae and Land Plants
4.2. Divergent Homologs to Fungal Mitoviruses Detected in Ostreobium sp.
4.3. Detection of Plant Viruses in the Chlorarachniophytes
4.4. First Report of a Negative-Sense RNA Virus in Microalgae
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.S.; Rise, M.L.; Culley, A.I.; Steward, G.F. RNA viruses in the sea. FEMS Microbiol. Rev. 2009, 33, 295–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; De Vargas, C.; Gasol, J.M.; et al. Patterns and ecological drivers of ocean viral communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Steward, G.F.; Culley, A.I.; Mueller, J.A.; Wood-Charlson, E.M.; Belcaid, M.; Poisson, G. Are we missing half of the viruses in the ocean? ISME J. 2013, 7, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Miranda, J.A.; Culley, A.I.; Schvarcz, C.R.; Steward, G.F. RNA viruses as major contributors to Antarctic virioplankton. Environ. Microbiol. 2016, 18, 3714–3727. [Google Scholar] [CrossRef]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using metagenomics to characterize an expanding virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.A.L.; Andrade, A.C.; Dos, S.P.; Boratto, P.V.d.M.; Trindade, G.d.S.; Kroon, E.G.; Abrahão, J.S. An anthropocentric view of the virosphere-host relationship. Front. Microbiol. 2017, 8, 1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. Metagenomic analysis of coastal RNA virus communities. Science 2006, 312, 1795–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. The complete genomes of three viruses assembled from shotgun libraries of marine RNA virus communities. Virol. J. 2007, 4, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, S.M.; Staniewski, M.A.; Chaban, Y.V.; Long, A.M.; Wang, D. Diversity of viruses infecting eukaryotic algae. Curr. Issues Mol. Biol. 2020, 39, 29–62. [Google Scholar] [CrossRef] [Green Version]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking virus genomes with host taxonomy. Viruses 2016, 8, 666. [Google Scholar] [CrossRef]
- Mayer, J.A.; Taylor, F.J.R. A virus which lyses the marine nanoflagellate Micromonas pusilla. Nature 1979, 281, 299–301. [Google Scholar] [CrossRef]
- Brown, R.M. Algal viruses. Adv. Virus Res. 1972, 17, 243–277. [Google Scholar]
- Brussaard, C.P.D.; Martínez, J. Algal Bloom Viruses. Plant Viruses 2008, 2, 1–10. [Google Scholar]
- Leliaert, F.; Smith, D.R.; Moreau, H.; Herron, M.D.; Verbruggen, H.; Delwiche, C.F.; De Clerck, O. Phylogeny and molecular evolution of the green algae. CRC Crit. Rev. Plant Sci. 2012, 31, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Urayama, S.I.; Takaki, Y.; Nunoura, T. FLDS: A comprehensive DSRNA sequencing method for intracellular RNA virus surveillance. Microbes Environ. 2016, 31, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushegian, A.; Shipunov, A.; Elena, S.F. Changes in the composition of the RNA virome mark evolutionary transitions in green plants. BMC Biol. 2016, 14, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Illergard, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence—A study of structural response in protein cores. Proteins Struct. Funct. Bioinform. 2009, 77, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.H.; Grimes, J.M.; Stuart, D.I. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 2005, 15, 655–663. [Google Scholar] [CrossRef]
- Chen, J.; Guo, M.; Wang, X.; Liu, B. A comprehensive review and comparison of different computational methods for protein remote homology detection. Brief. Bioinform. 2018, 19, 231–244. [Google Scholar] [CrossRef]
- Singh, J.; Saxena, R.C. An introduction to microalgae: Diversity and significance. In Handbook of Marine Microalgae; Academic Press: Cambridge, MA, USA, 2015; pp. 11–24. ISBN 9780128007761. [Google Scholar]
- Archibald, J.M. The evolution of algae by secondary and tertiary endosymbiosis. Adv. Bot. Res. 2012, 64, 87–118. [Google Scholar]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G.B. The New Tree of Eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Richmond, A.; Hu, Q. Handbook of Microalgal Culture: Applied Phycology and Biotechnology, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 1–719. ISBN 9781118567166. [Google Scholar]
- Guiry, M.D. How many species of algae are there? J. Phycol. 2012, 48, 1057–1063. [Google Scholar] [CrossRef]
- Chapman, R.L. Algae: The world’s most important “plants”—an introduction. Mitig. Adapt. Strateg. Glob. Chang. 2013, 18, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.; Knoll, A.H.; Chan, C.X.; Verbruggen, H. Plastid phylogenomics with broad taxon sampling further elucidates the distinct evolutionary origins and timing of secondary green plastids. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Prangishvili, D. The major role of viruses in cellular evolution: Facts and hypotheses. Curr. Opin. Virol. 2013, 3, 558–565. [Google Scholar] [CrossRef]
- Wetherbee, R.; Marcelino, V.R.; Costa, J.F.; Grant, B.; Crawford, S.; Waller, R.F.; Andersen, R.A.; Berry, D.; McFadden, G.I.; Verbruggen, H. A new marine prasinophyte genus alternates between a flagellate and a dominant benthic stage with microrhizoids for adhesion. J. Phycol. 2019, 55, 1210–1225. [Google Scholar] [CrossRef]
- Wetherbee, R.; Verbruggen, H. Kraftionema allantoideum, a new genus and family of Ulotrichales (Chlorophyta) adapted for survival in high intertidal pools. J. Phycol. 2016, 52, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Marcelino, V.R.; Clausen, P.T.L.C.; Buchmann, J.P.; Wille, M.; Iredell, J.R.; Meyer, W.; Lund, O.; Sorrell, T.C.; Holmes, E.C. CCMetagen: Comprehensive and accurate identification of eukaryotes and prokaryotes in metagenomic data. Genome Biol. 2020, 21, 103. [Google Scholar] [CrossRef]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Vieira, H.H.; Bagatini, I.L.; Guinart, C.M.; Vieira, A.A.H.; Vieira, H.H.; Bagatini, I.L.; Guinart, C.M.; Vieira, A.A.H. tufA gene as molecular marker for freshwater Chlorophyceae. ALGAE 2016, 31, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Strassert, J.F.H.; Jamy, M.; Mylnikov, A.P.; Tikhonenkov, D.V.; Burki, F. New phylogenomic analysis of the enigmatic phylum telonemia further resolves the eukaryote tree of life. Mol. Biol. Evol. 2019, 36, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Cortona, A.; Jackson, C.J.; Bucchini, F.; Van Bel, M.; D’hondt, S.; Skaloud, P.; Delwiche, C.F.; Knoll, A.H.; Raven, J.A.; Verbruggen, H.; et al. Neoproterozoic origin and multiple transitions to macroscopic growth in green seaweeds. Proc. Natl. Acad. Sci. USA 2020, 117, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Cocquyt, E.; Verbruggen, H.; Leliaert, F.; Clerck, O. De Evolution and cytological diversification of the green seaweeds (Ulvophyceae). Mol. Biol. Evol. 2010, 27, 2052–2061. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA polymerases: Insights from structure, function and evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E. V Origins and evolution of the global RNA virome. mBio 2018, 9, e02318–e02329. [Google Scholar] [CrossRef] [Green Version]
- Koga, R.; Horiuchi, H.; Fukuhara, T. Double-stranded RNA replicons associated with chloroplasts of a green alga, Bryopsis cinicola. Plant Mol. Biol. 2003, 51, 991–999. [Google Scholar] [CrossRef]
- Le Alsumard, T.C.; Golubic, S.; Priess, K. Fungi in corals: Symbiosis or disease? Interaction between polyps and fungi causes pearl-like skeleton biomineralization. Mar. Ecol. Prog. Ser. 1995, 117, 137–148. [Google Scholar] [CrossRef]
- Sabanadzovic, S.; Valverde, R.A.; Brown, J.K.; Martin, R.R.; Tzanetakis, I.E. Southern tomato virus: The link between the families Totiviridae and Partitiviridae. Virus Res. 2009, 140, 130–137. [Google Scholar] [CrossRef]
- Koga, R.; Fukuhara, T.; Nitta, T. Molecular characterization of a single mitochondria-associated double-stranded RNA in the green alga Bryopsis. Plant Mol. Biol. 1998, 36, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef] [Green Version]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Plant viruses of the Amalgaviridae family evolved via recombination between viruses with double-stranded and negative-strand RNA genomes. Biol. Direct 2015, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Tomaru, Y.; Shimabukuro, H.; Kimura, K.; Hirai, M.; Takaki, Y.; Hagiwara, D.; Takuro, N.; Urayma, S.-I. Viral RNA genomes identified from marine macroalgae and a diatom. Microbes Environ. 2020, 35. [Google Scholar] [CrossRef]
- Dinan, A.M.; Lukhovitskaya, N.I.; Olendraite, I.; Firth, A.E. A case for a negative-strand coding sequence in a group of positive-sense RNA viruses. Virus Evol. 2020, 6, veaa007. [Google Scholar]
- Waldron, F.M.; Stone, G.N.; Obbard, D.J. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet. 2018, 14, e1007533. [Google Scholar] [CrossRef] [Green Version]
- Polischuk, V.; Budzanivska, I.; Shevchenko, T.; Oliynik, S. Evidence for plant viruses in the region of Argentina Islands, Antarctica. FEMS Microbiol. Ecol. 2007, 59, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Petrzik, K.; Vondrák, J.; Kvíderová, J.; Lukavský, J. Platinum anniversary: Virus and lichen alga together more than 70 years. PLoS ONE 2015, 10, e01207688. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.J.; Park, D.; Lee, J.S.; Sebastiani, F.; Hahn, Y. Identification of a novel plant amalgavirus (Amalgavirus, Amalgaviridae) genome sequence in Cistus incanus. Acta Virol. 2018, 62, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhan, B.; Cao, M.; Wang, K.; Wang, X.; Zhou, X. Detection and characterization of cucumis melo cryptic virus, cucumis melo amalgavirus 1, and melon necrotic spot virus in Cucumis melo. Viruses 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Goh, C.J.; Park, D.; Hahn, Y. Identification of a novel plant RNA virus species of the genus Amalgavirus in the family Amalgaviridae from chia (Salvia hispanica). Genes Genom. 2019, 41, 507–514. [Google Scholar] [CrossRef]
- Roossinck, M.J. Lifestyles of plant viruses. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1899–1905. [Google Scholar] [CrossRef] [Green Version]
- Nibert, M.L.; Ghabrial, S.A.; Maiss, E.; Lesker, T.; Vainio, E.J.; Jiang, D.; Suzuki, N. Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res. 2014, 188, 128–141. [Google Scholar] [CrossRef]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef]
- Roossinck, M.J. Evolutionary and ecological links between plant and fungal viruses. New Phytol. 2019, 221, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Bonfante, P. Algae and fungi move from the past to the future. eLife 2019, 8, e49448. [Google Scholar] [CrossRef]
- Ricci, F.; Marcelino, V.R.; Blackall, L.L.; Kühl, M.; Medina, M.; Verbruggen, H. Beneath the surface: Community assembly and functions of the coral skeleton microbiome. Microbiome 2019, 7, 1–10. [Google Scholar] [CrossRef]
- Raghukumar, C.; Ravindran, J. Fungi and Their Role in Corals and Coral Reef Ecosystems. In Biology of Marine Fungi; Raghukumar, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 89–113. [Google Scholar]
- Marcelino, V.R.; Verbruggen, H. Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Sci. Rep. 2016, 6, 31508. [Google Scholar] [CrossRef] [PubMed]
- Nerva, L.; Vigani, G.; Di Silvestre, D.; Ciuffo, M.; Forgia, M.; Chitarra, W.; Turina, M. Biological and molecular characterization of chenopodium quinoa mitovirus 1 reveals a distinct small RNA response compared to those of cytoplasmic RNA viruses. J. Virol. 2019, 93, e01918–e01998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charon, J.; Grigg, M.J.; Eden, J.-S.; Piera, K.A.; Rana, H.; William, T.; Rose, K.; Davenport, M.P.; Anstey, N.M.; Holmes, E.C. Novel RNA viruses associated with Plasmodium vivax in human malaria and Leucocytozoon parasites in avian disease. PLoS Pathog. 2019, 15, e1008216. [Google Scholar] [CrossRef] [Green Version]
- Zangger, H.; Ronet, C.; Desponds, C.; Kuhlmann, F.M.; Robinson, J.; Hartley, M.-A.; Prevel, F.; Castiglioni, P.; Pratlong, F.; Bastien, P.; et al. Detection of Leishmania RNA virus in Leishmania parasites. PLoS Negl. Trop. Dis. 2013, 7, e2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grybchuk, D.; Akopyants, N.S.; Kostygov, A.Y.; Konovalovas, A.; Lye, L.-F.; Dobson, D.E.; Zangger, H.; Fasel, N.; Butenko, A.; Frolov, A.O.; et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl. Acad. Sci. USA 2018, 115, E506–E515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, G.; Myers, K.; Fry, W.E.; Hillman, B.I. A member of the virus family Narnaviridae from the plant pathogenic oomycete Phytophthora infestans. Arch. Virol. 2012, 157, 165–169. [Google Scholar] [CrossRef]
- Yoon, H.S.; Hackett, J.D.; Ciniglia, C.; Pinto, G.; Bhattacharya, D. A molecular timeline for the origin of photosynthetic eukaryotes. Mol. Biol. Evol. 2004, 21, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Parfrey, L.W.; Lahr, D.J.G.; Knoll, A.H.; Katz, L.A. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl. Acad. Sci. USA 2011, 108, 13624–13629. [Google Scholar] [CrossRef] [Green Version]
- Burki, F.; Keeling, P.J. Rhizaria. Curr. Biol. 2014, 24, R103–R107. [Google Scholar] [CrossRef] [Green Version]
- Sven, B. Gould Algae’s complex origins. Nature 2012, 492, 46–48. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Species | Class/Family |
|---|---|---|
| ALG_1 | Kraftionema allantoideum | Ulvophyceae/Kraftionemaceae |
| Microrhizoidea pickettheapsiorum | Mamiellophyceae/Dolichomastigaceae | |
| Dolichomastix tenuilepis | Mamiellophyceae/Dolichomastigaceae | |
| ALG_2 | Ostreobium sp. HV05007bc | Ulvophyceae/Bryopsidales |
| ALG_3 | Chlorarachnion reptans | Chlorarachniophyceae/Chlorarachnion |
| Lotharella sp | Chlorarachniophyceae/Lotharella |
| New Virus (Algal Host Species) | Length nt | PE Read Count (% Non rRNA) | BLASTx Hit GenBank Acc. | %ID | e-Value | BLASTx Hit Organism | BLASTx Hit Taxonomy |
|---|---|---|---|---|---|---|---|
| Amalga-like boulavirus (K. allantoideum) | 1440 | 4313 (0.17%) | BAA25883 | 31 | 4.6 × 1023 | BDRM | Partitiviridae (dsRNA) |
| Amalga-like chassivirus (Ostreobium sp.) | 3399 | 1503 (0.02%) | BAA25883 | 28 | 1.8 × 1038 | BDRM | Partitiviridae (dsRNA) |
| Amalga-like chaucrivirus (Ostreobium sp.) | 4036 | 16,934 (0.23%) | BAA25883 | 33 | 3.3 × 10103 | BDRM | Partitiviridae (dsRNA) |
| Amalga-like dominovirus (Ostreobium sp.) | 4011 | 2996 (0.04%) | BAA25883 | 33 | 5.1 × 1088 | BDRM | Partitiviridae (dsRNA) |
| Amalga-like lacheneauvirus (Ostreobium sp.) | 3254 | 934 (0.01%) | BAA25883 | 27 | 3.5 × 1039 | BDRM | Partitiviridae (dsRNA) |
| Partiti-like alassinovirus (Ostreobium sp.) | 3658 | 5135 (0.07%) | BAB63954 | 29 | 1.6 × 1048 | BDRC | Bryopsis cinicola * (dsRNA) |
| Partiti-like lacotivirus (Ostreobium sp.) | 3273 | 92,840 (1.26%) | BAB63954 | 29 | 2.5 × 1045 | BDRC | Bryopsis cinicola * (dsRNA) |
| Partiti-like adriusvirus (Ostreobium sp.) | 4252 | 4833 (0.07%) | BAB63954 | 23 | 5.7 × 1018 | BDRC | Bryopsis cinicola * (dsRNA) |
| Mito-like babylonusvirus (Ostreobium sp.) | 2942 | 9029 (0.12%) | APG77166 | 38 | 9 × 1042 | Shahe narna-like virus 6 | Unclassified RNA virus (ssRNA) |
| Mito-like albercanusvirus (Ostreobium sp.) | 2791 | 5294 (0.07%) | APG77166 | 39 | 7.2 × 1041 | Shahe narna-like virus 6 | Unclassified RNA virus (ssRNA) |
| Mito-like spartanusvirus (Ostreobium sp.) | 2684 | 15,388 (0.21%) | ASM94099 | 38 | 2 × 1032 | Barns Ness serrated wrack narna-like virus 3 | Narnaviridae (ss+RNA) |
| Mito-like laruketanusvirus (Ostreobium sp.) | 2928 | 14,185 (0.19%) | APG77166.1 | 36 | 5.8 × 1033 | Shahe narna-like virus 6 | Unclassified RNA virus (ssRNA) |
| Mito-like bobnusvirus (Ostreobium sp.) | 2773 | 2621 (0.04%) | APG77166 | 34 | 6.1 × 1040 | Shahe narna-like virus 6 | Unclassified RNA virus |
| Mito-like picolinusvirus (Ostreobium sp.) | 2714 | 8792 (0.12%) | YP 00228433 | 34 | 4.1 × 1033 | Botrytis cinerea mitovirus 1 | Narnaviridae (ss+RNA) |
| Mito-like bionusvirus (Ostreobium sp.) | 3260 | 7529 (0.10%) | AXY40442 | 27 | 5.7 × 1013 | Rhizophagus diaphanum mitovirus 1 | Narnaviridae (ss+RNA) |
| Tombus-like chagrupourvirus (Ostreobium sp.) | 3835 | 5418 (0.07%) | YP 009336735 | 36 | 5.1 × 1045 | Hubei tombus-like virus 12 | Unclassified RNA virus |
| Virga-like bellevillovirus (C. reptans) | 2313 | 229 (0.01%) | AMO03254 | 29 | 4.4 × 1044 | Boutonnet virus | Unclassified ssRNA virus (ssRNA) |
| Bunya-like bridouvirus (C. reptans) | 2818 | 208 (0.01%) | APG79310 | 30 | 9.1 × 1068 | Shahe bunya-like virus 1 | Unclassified RNA virus |
| Contig Name | ORF | Abund. | Viral Hit | e-Value | Viral Hit Description | Viral-Like Hit Taxonomy | PFAM Hit ID | PFAM e-Value | PFAM Hit Description |
|---|---|---|---|---|---|---|---|---|---|
| ALG_2_DN19089_c0_g1_i1_len4252 | ORF_1 | 4717 | VOG03062 | 1.00 × 1012 | REFSEQ hypothetical protein | Bacteriophage | - | - | - |
| ALG_2_DN19089_c0_g1_i1_len4252 | ORF_1 | 4717 | PF00680.20 | 2.70 × 105 | RNA dependent RNA polymerase | RdRP-1 | - | - | - |
| ALG_2_DN19250_c2_g3_i5_len1869 | ORF_1 | 743.91 | VOG23558 | 1.00 × 104 | REFSEQ hypothetical protein | Caudovirales; Siphoviridae | - | - | - |
| ALG_2_DN18568_c0_g1_i1_len2977 | ORF_1 | 511 | VOG10478 | 1.30 × 106 | sp|Q05224|VG18 BPML5 Gene 18 protein | Bacteriophage | - | - | - |
| ALG_2_DN19013_c0_g1_i2_len1689 | ORF_2 | 224.31 | VOG22975 | 1.40 × 104 | REFSEQ carboxylesterase | Caudovirales; Siphoviridae | - | - | - |
| ALG_2_DN19410_c0_g2_i6_len1950 | ORF_1 | 183.94 | VOG12013 | 5.20 × 104 | sp|P03778|Y06 BPT7 Protein 0.6B | Viruses | PF16752.5 | 1.50 × 104 | Tubulin-specific chaperone C |
| ALG_2_DN18226_c0_g1_i1_len1259 | ORF_1 | 157.61 | VOG09820 | 2.90 × 104 | REFSEQ hypothetical protein | Phycodnaviridae; Chlorovirus | - | - | - |
| ALG_2_DN18993_c2_g2_i2_len2532 | ORF_1 | 151.17 | VOG08344 | 8.50 × 108 | REFSEQ hypothetical protein | Bacteriophage | PF13424.6 | 3.80 × 10132 | Tetratricopeptide repeat |
| ALG_3_DN34624_c0_g1_i1_len2077 | ORF_2 | 146 | PF02123.16 | 8.50 × 105 | Viral RNA-directed RNA-polymerase | RdRP-4 | - | - | - |
| ALG_2_DN18744_c0_g1_i3_len2080 | ORF_1 | 134.99 | VOG10472 | 4.10 × 104 | REFSEQ hypothetical protein | Poxviridae | - | - | - |
| ALG_2_DN19214_c2_g1_i7_len1432 | ORF_2 | 118 | VOG06927 | 6.90 × 104 | REFSEQ hypothetical protein | Bacteriophage | - | - | - |
| ALG_3_DN25592_c0_g1_i1_len1043 | ORF_1 | 115 | VOG01256 | 4.40 × 104 | sp|Q9QU29|ORF3 TTVB1 Uncharacterized ORF3 protein | dsDNA viruses | - | - | - |
| ALG_2_DN18451_c0_g1_i4_len2061 | ORF_4 | 109.48 | VOG17696 | 7.10 × 104 | REFSEQ hypothetical protein | Bacteriophage | PF16058.5 | 1.80 × 1017 | Mucin-like |
| ALG_2_DN18451_c0_g1_i4_len2061 | ORF_4 | 109.48 | VOG17696 | 7.10 × 104 | REFSEQ_hypothetical_protein | Bacteriophage | PF16058.5 | 1.10 × 107 | Mucin-like |
| ALG_2_DN18732_c0_g2_i2_len2454 | ORF_2 | 99.65 | VOG09815 | 1.70 × 1015 | REFSEQ hypothetical protein | Phycodnaviridae; Chlorovirus | - | - | - |
| ALG_2_DN18957_c0_g1_i1_len1408 | ORF_2 | 97.98 | VOG02199 | 8.40 × 104 | sp|Q5UR09|YR648 MIMIV Uncharacterized protein R648 | Ortervirales | PF06156.13 | 4.70 × 104 | Initiation control protein YabA |
| ALG_2_DN18957_c0_g1_i2_len1657 | ORF_2 | 89.04 | VOG02199 | 3.50 × 104 | sp|Q5UR09|YR648 MIMIV Uncharacterized protein R648 | Ortervirales | PF06156.13 | 2.20 × 104 | Initiation control protein YabA |
| ALG_2_DN19250_c2_g3_i3_len2203 | ORF_1 | 71.48 | VOG23558 | 7.80 × 105 | REFSEQ hypothetical protein | Caudovirales; Siphoviridae | - | - | - |
| ALG_2_DN19410_c0_g2_i7_len1634 | ORF_1 | 49.51 | VOG12013 | 9.90 × 104 | sp|P03778|Y06 BPT7 Protein 0.6B | Bacteriophage | - | - | - |
| ALG_2_DN11543_c0_g1_i1_len842 | ORF_1 | 42 | VOG20356 | 3.80 × 104 | REFSEQ hypothetical protein | Caudovirales; Myoviridae | - | - | - |
| ALG_2_DN19463_c5_g2_i1_len765 | ORF_1 | 37.7 | VOG24589 | 2.60 × 105 | REFSEQ hypothetical protein | Caudovirales; Siphoviridae | - | - | - |
| ALG_2_DN19174_c0_g2_i18_len938 | ORF_1 | 36.39 | VOG10625 | 9.70 × 104 | sp|Q05293|VG78 BPML5 Gene 78 protein | Bacteriophage | - | - | - |
| ALG_2_DN41289_c0_g1_i1_len750 | ORF_1 | 36 | VOG06662 | 9.50 × 105 | REFSEQ Cupin | Bacteriophage | - | - | - |
| ALG_2_DN22182_c0_g1_i1_len820 | ORF_1 | 35 | VOG02199 | 1.50 × 104 | sp|Q5UR09|YR648 MIMIV Uncharacterized protein R648 | Ortervirales | PF06156.13 | 7.20 × 104 | Initiation control protein YabA |
| ALG_2_DN44027_c0_g1_i1_len815 | ORF_1 | 33.98 | VOG21678 | 3.70 × 104 | REFSEQ hypothetical protein | Caudovirales; Myoviridae | PF08614.11 | 6.00 × 105 | Autophagy protein 1(ATG16) |
| ALG_2_DN594_c0_g2_i1_len711 | ORF_1 | 28 | PF17501.2 | 2.80 × 104 | Viral RNA-directed RNA polymerase | Viral_RdRp_C | - | - | - |
| ALG_2_DN14271_c0_g1_i1_len772 | ORF_1 | 25.12 | VOG18617 | 2.20 × 104 | REFSEQ hypothetical protein | Caudovirales; Siphoviridae | PF13855.6 | 1.60 × 1021 | Leucine rich repeat |
| ALG_2_DN18993_c2_g2_i1_len2178 | ORF_1 | 21.18 | VOG08344 | 4.70 × 107 | REFSEQ hypothetical protein | Bacteriophage | PF13374.6 | 1.40 × 10126 | 1Tetratricopeptide repeat |
| ALG_2_DN44027_c0_g1_i2_len783 | ORF_1 | 19.02 | VOG21678 | 3.60 × 104 | REFSEQ hypothetical protein | Caudovirales; Myoviridae | PF08614.11 | 7.70 × 105 | Autophagy protein 1(ATG16) |
| ALG_2_DN19463_c5_g2_i6_len928 | ORF_1 | 14.34 | VOG24589 | 6.40 × 105 | REFSEQ hypothetical protein | Caudovirales; Siphoviridae | - | - | - |
| ALG_2_DN19463_c5_g2_i4_len863 | ORF_1 | 4.36 | VOG24589 | 5.30 × 105 | REFSEQ hypothetical protein | Caudovirales; Siphoviridae | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charon, J.; Marcelino, V.R.; Wetherbee, R.; Verbruggen, H.; Holmes, E.C. Metatranscriptomic Identification of Diverse and Divergent RNA Viruses in Green and Chlorarachniophyte Algae Cultures. Viruses 2020, 12, 1180. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101180
Charon J, Marcelino VR, Wetherbee R, Verbruggen H, Holmes EC. Metatranscriptomic Identification of Diverse and Divergent RNA Viruses in Green and Chlorarachniophyte Algae Cultures. Viruses. 2020; 12(10):1180. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101180
Chicago/Turabian StyleCharon, Justine, Vanessa Rossetto Marcelino, Richard Wetherbee, Heroen Verbruggen, and Edward C. Holmes. 2020. "Metatranscriptomic Identification of Diverse and Divergent RNA Viruses in Green and Chlorarachniophyte Algae Cultures" Viruses 12, no. 10: 1180. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101180